
Electrochemical oxygen activation driven by enamine-keto push–pull interactions in triazole-based covalent organic polymers†
Surajit
Samui‡
a,
Greesh
Kumar‡
a,
Thakur Rochak Kumar
Rana
b and
Ramendra Sundar
Dey
 *a
*a
aInstitute of Nano Science and Technology, Sector-81, Mohali 140306, Punjab, India. E-mail: rsdey@inst.ac.in
bDepartment of Chemistry, Indian Institute Institute of Technology, Powai, Mumbai-400076, India
First published on 6th November 2024
Abstract
We have developed two triazole-based covalent organic polymers (COPs) with donor–acceptor motifs. The keto-enriched COP demonstrated exceptional oxygen activation via electrochemical stimuli, driven by strong push–pull interactions. In situ studies and DFT calculations confirmed the critical role of enamine carbon positive charges in enhancing performance, setting new benchmarks in COP design.
The electrochemical oxygen reduction reaction (ORR) is vital for fuel cells and rechargeable metal–air batteries (MABs), involving complex proton-coupled electron transfers to break the O
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, resulting in slow kinetics.1,2 To accelerate the ORR process, noble metal-based electrocatalysts like Pt/C and Pd/C, have traditionally been employed due to their strong oxygen-binding properties. However, these catalysts come with notable drawbacks, including high cost, scarcity, and poor long-term stability, which limit their practical utility.3,4 Consequently, substantial efforts have been made to develop non-precious metal catalysts. Despite their potential, performance heavily relies on the design of active sites, porosity, and heteroatom composition.5 The limitations of noble metals and transition metal-based catalysts have driven interest in the search for metal-free catalysts that offer both precise active sites and long-term stability.
O bond, resulting in slow kinetics.1,2 To accelerate the ORR process, noble metal-based electrocatalysts like Pt/C and Pd/C, have traditionally been employed due to their strong oxygen-binding properties. However, these catalysts come with notable drawbacks, including high cost, scarcity, and poor long-term stability, which limit their practical utility.3,4 Consequently, substantial efforts have been made to develop non-precious metal catalysts. Despite their potential, performance heavily relies on the design of active sites, porosity, and heteroatom composition.5 The limitations of noble metals and transition metal-based catalysts have driven interest in the search for metal-free catalysts that offer both precise active sites and long-term stability.
Recently, polymer materials such as covalent organic polymers (COPs),6 covalent triazine frameworks (CTFs)7 and covalent organic frameworks (COFs),8,9 have gained significant attention as promising catalysts for the oxygen reduction reaction (ORR), due to their high structural precision, tunable porosity, and chemical stability. Among these, COPs have been explored for ORR applications owing to their modular design, which allows precise control over electro-active sites, surface area, and pore size, providing a versatile platform for catalysis.10,11
Despite the shared advantages of covalent organic polymers (COPs) for ORR applications, these materials face certain challenges that affect their catalytic efficiency. One major limitation is the difficulty in optimizing the active sites to enhance electron transport and oxygen binding efficiency. The performance of these materials is highly influenced by factors such as electron density, molecular orientation, bonding angles of their building blocks, and the availability of heteroatoms, all of which determine the nature and accessibility of the active sites.12,13 Another important aspect for the ORR process is the selectivity of the pathway for a particular material. Two electron pathway mediated production of H2O2 is not desirable for fuel cell and Zn–air battery applications as compared to the four electron pathway mediated generation of H2O molecules as it leads to corrosion and lowering of the efficiency of the electrocatalyst. Therefore, further fine-tuning of these parameters such as. (a) active sites, (b) the electron transfer rate, and (c) the stability of the framework material, are necessary to fully unlock the potential of COP materials in ORR catalysis. In triazole-based COPs, the N-rich triazole unit can induce high electron density, which is beneficial for the electrocatalytic ORR; however, the chemistry of these active sites is still not fully understood, offering significant potential for further research. Dai et al.14 attributed ORR enhancement to nitrogen-induced charge delocalization, which shifts O2 adsorption from the usual end-on mode (Pauling model) to a side-on mode (Yeager model), weakening the O–O bond and thus improving the reaction. Therefore, constructing donor–acceptor entangled COPs with predefined active sites, along with a thorough investigation of catalytic activity supported by theoretical calculations, could lead to significant findings. Although there are several studies in the literature regarding the strategic design and engineering of COPs, but effective utilization of each constituent monomer unit for the electrochemical ORR process and the comprehensive understanding of the relationship between catalyst structure and activity is still an area to be explored.
In this study, we strategically synthesized two COP materials designated as RT-COP and PT-COP by combining 2,4-dihydroxy-1,3,5-triformylcarbaldehyde (DHTA) and 2-hydroxy-1,3,5-triformylcarbaldehyde (HTA) with 3,5-diamino-1,2,4-triazole, respectively. The detailed syntheses of the RT-COP and PT-COP materials and their monomer units are provided in ESI,† Section S1. The synthesized catalysts with potential active sites were explored in electrochemical ORR catalysis in alkaline media. The reaction kinetics and intermediates evolved during the ORR were analyzed using in situ FTIR studies. Density functional theory (DFT) confirmed that the strategically molded carbon site of β-ketoenamine effectively catalyzed the adsorption and reduction of O2 to water. The strategically incorporated β-ketoenamine groups in PT-COP and RT-COP induce positive charges, as illustrated in Scheme 1. However, RT-COP, which contains a greater number of β-ketoenamine groups, exhibits a stronger positive charge on the enamine carbon, leading to optimal binding strength and enhanced push–pull interactions for the subsequent reduction process. The favourable binding affinity of oxygen to RT-COP was further evaluated through DFT, where O2 binding to the COPs was found to follow the energetically more favourable Yeager model via end-on adsorption. The presence of more β-keto groups in RT-COP ensures stronger enamine (C–NH) interactions by generating a higher δ+ charge through the inductive effect, which results in improved adsorption and polarization of O2.
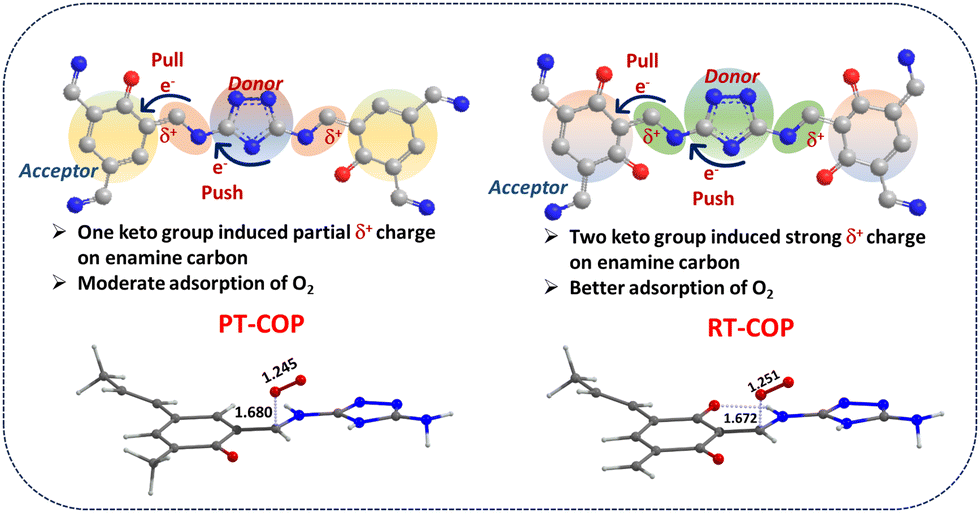 | ||
| Scheme 1 Diagrammatic representation of push–pull interaction present in COPs followed by oxygen binding following the Yeager model presented in the study. | ||
Powder X-ray diffraction (PXRD) analysis of the RT-COP and PT-COP catalyst confirms amorphous nature with 2θ values of 27° indicating π–π stacking in both the materials (Fig. S1, ESI†). Solid state 13C cross polarization magic angle spinning (CP-MAS) NMR confirmed the presence of β-ketoenamine and an imine bond in both the materials (Fig. 1(a) and Fig. S2, ESI†). Bonding connectivity, as confirmed by Fourier-transform infrared spectroscopy (FTIR), indicated the presence of a keto group and CC bond of β-ketoenamine and imine bond in RT-COP, denoted by stretching frequencies at 1633 cm−1, 1437 cm−1 and 1576 cm−1, respectively (Fig. 1(b)). In contrast, PT-COP exhibited stretching frequencies at 1618 cm−1 and 1588 cm−1, confirming the presence of keto–enol tautomerism as well as imine bonds.15–17 A broad stretching frequency around ∼3380 cm−1 for both covalent organic polymers confirmed the presence of N–H stretching, further supporting the formation of a tautomerized structure. Morphological analysis of RT-COP (Fig. 1(c)) and PT-COP (Fig. S3a–c and S4a, b, ESI†) conducted through field emission scanning electron microscopy (FESEM) confirmed the presence of agglomerated particles. Energy dispersive X-ray spectroscopy (EDS) demonstrated a homogeneous distribution of carbon (C), nitrogen (N), and oxygen (O) elements throughout the materials (Fig. S3d–f, ESI†). Furthermore, elemental composition and chemical states were confirmed through X-ray photoelectron spectroscopy (XPS) (Fig. 1(d), and Fig. S5a, b, ESI†). The high-resolution C 1s spectrum of RT-COP exhibited peaks at 284.5 eV, 286.26 eV, 287.4 eV, and 289.35 eV, confirming the presence of CC, CN, C–N, and CO functionalities, respectively (Fig. 1(d)).18 Similarly, deconvoluted peaks of the N 1s spectrum showed the peaks at 398.5 eV, 399.5 eV, and 400.7 eV confirming the CN, C–N and N–H functionality, respectively (Fig. 1(d)). The presence of CO and O–H functionality was also confirmed through the high resolution XPS spectrum of O 1s (Fig. S5b, ESI†) at 530.6 eV and 532.5 eV, respectively.16 Similarly, the high resolution C 1s spectrum of PT-COP (Fig. S6a–d, ESI†) confirmed CC, CN, C–N and CO functionality through deconvoluted peaks at 284.8 eV, 286.2 eV, 287.3 eV, and 289 eV. The deconvoluted N 1s spectrum of PT-COP confirmed the –NH, C–N, and CN functionality from the binding energies at 400.4 eV, 399.17 eV, and 398.5 eV. The elemental composition of both materials is summarized in ESI,† Table T1.
 | ||
| Fig. 1 (a) Solid state 13C CP-MAS NMR data of RT-COP. (b) FTIR stretching frequency of RT-COP and PT-COP. (c) FESEM image of RT-COP. (d) High-resolution XPS spectrum of C 1s and N 1s for the RT-COP catalyst. | ||
The electrocatalytic activity for the oxygen reduction reaction (ORR) of RT-COP and PT-COP materials was assessed using cyclic voltammetry (CV) and linear sweep voltammetry (LSV) in a 0.1 M KOH solution. CV measurements revealed a sharp cathodic peak under oxygen-saturated conditions for both materials, confirming their ORR activity, which was absent in argon-saturated solutions (Fig. S7, ESI†). The optimal ratio of activated carbon (acetylene black) to RT-COP was also determined (Fig. S8, ESI†), with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio being the most effective and used for further analyses in this work. Additional studies using a rotating ring-disk electrode (RRDE) demonstrated that RT-COP exhibited enhanced performance, with a half-wave potential (E1/2) of 0.76 V vs. RHE and a limiting current density (J1) of 4.4 mA cm−2, which is higher than that of PT-COP (E1/2 = 0.72 V vs. RHE, J1 = 3.9 mA cm−2) but inferior to that of the standard Pt/C (20 wt%) catalyst (Fig. S9, ESI†).
:1 ratio being the most effective and used for further analyses in this work. Additional studies using a rotating ring-disk electrode (RRDE) demonstrated that RT-COP exhibited enhanced performance, with a half-wave potential (E1/2) of 0.76 V vs. RHE and a limiting current density (J1) of 4.4 mA cm−2, which is higher than that of PT-COP (E1/2 = 0.72 V vs. RHE, J1 = 3.9 mA cm−2) but inferior to that of the standard Pt/C (20 wt%) catalyst (Fig. S9, ESI†).
The kinetics of the oxygen reduction reaction (ORR) were analyzed using a Tafel plot derived from polarization curves. RT-COP exhibited a lower Tafel slope of 64 mV dec−1 compared to PT-COP (72 mV dec−1) and Pt/C (77 mV dec−1), indicating faster ORR kinetics (Fig. 2(b)). All electrochemical parameters for the ORR are summarized in ESI,† Table T2. The electrochemical active surface area (ECSA) of RT-COP was calculated to be 1.49 cm2, higher than PT-COP (1.42 cm2), suggesting more abundant active sites and better electron transport (Fig. S10a–d, ESI†). Electrochemical impedance spectroscopy (EIS) supported this, as RT-COP exhibited lower series resistance (Rs) and charge-transfer resistance (Rct) (Fig. S11a and b, ESI†) reflecting better electron transportation on the catalyst surface during the ORR process. The LSV measurements at various rotation speeds (Fig. S12a and b, ESI†) further clarified the ORR mechanism, demonstrating enhanced limiting current density with increasing rotation speed, indicating improved mass transport of oxygen. The Koutecky–Levich (K–L) plot for RT-COP and PT-COP showed good linearity as shown in Fig. 2(c) and Fig. S13, ESI,† confirming first-order reaction kinetics towards the ORR.19 The RRDE technique (Fig. S14a and b, ESI†) was used to estimate the number of electrons involved in the ORR process,20 with RT-COP and PT-COP materials following a nearly 4-electron pathway (n = 3.99 for RT-COP, 3.15 for PT-COP), leading to the direct conversion of O2 to water (inset Fig. 2(c)). Importantly, the H2O2 yield remained below 1%, further verifying the 4-electron pathway, making RT-COP suitable for practical applications like fuel cells, where minimal peroxide formation is critical.
 | ||
| Fig. 2 ORR electrochemical performance of RT-COP and other controlled samples in 0.1 M KOH solution. (a) LSV polarization curves of RT-COP, and PT-COP. (b) Tafel slope of RT-COP and other controlled catalysts. (c) Koutecky–Levich (K–L) plots of RT-COP at various potentials (inset: no of electrons and H2O2 yield involve during the ORR process with RT-COP and PT-COP catalysts). (d) Chronoamperometry response of RT-COP and Pt/C for ORR in alkaline media. | ||
The stability of RT-COP was tested using chronoamperometric measurements at a constant potential of 0.57 V vs RHE, showing that the material retained 91% of its initial current after 12 hours (Fig. 2(d)), outperforming the Pt/C catalyst, which showed significant degradation due to metal leaching in alkaline media. The stability was also confirmed by minimal shifts in the half-wave potential (only 7 mV) before and after testing (Fig. S15, ESI†), as well as by XPS analysis, which revealed consistent C1s, N1s and O1s spectra before and after stability testing (Fig. S16a–c, ESI†). Additionally, the methanol tolerance of RT-COP was evaluated through chronoamperometry in the presence of 1 M CH3OH in O2-saturated KOH solution (Fig. S17, ESI†). RT-COP showed no significant change in current density, indicating strong resistance to methanol crossover, a common issue in fuel cells. In contrast, Pt/C exhibited methanol oxidation, which interfered with its ORR performance. The RT-COP demonstrates excellent ORR performance, stability, and methanol tolerance, making it a promising candidate for fuel cell applications. Its superior ORR kinetics, large number of active sites, high electrochemically active surface area (ECSA), efficient electron transport, and low H2O2 yield all contribute to its robust electrocatalytic performance.
The multiple electron-proton coupled pathways of the oxygen reduction reaction (ORR) can generate H2O2 through a two-electron process and H2O through a four-electron process. While product selectivity can be determined through rotating ring-disk electrode (RRDE) experiments, mechanistic investigations of the process remain underexplored. To identify the intermediates evolved during the ORR process, in situ FTIR spectroscopy was conducted. The electrochemically stimulated ORR process initiates with the adsorption of the reactant O2 molecule onto the electrophilic carbon site of the enamine group, as indicated by the in situ FT-IR data presented in Fig. 3(a) at a stretching frequency of 1045 cm−1. The further reduction of the adsorbed oxygen molecule, accompanied by the subsequent addition of electrons and protons, was confirmed by the stretching frequency observed at 1392 cm−1, indicating the evolution of the O2− intermediate. Additionally, the stretching frequency at 1530 cm−1, denoting the bending motion of the H–O–H bond, confirmed the formation of water as the final product. With an increase in potential, a gradual rise in the broad stretching frequency between 3400 and 3500 cm−1 indicated the formation of H2O as the major product.21 But all the intermediate peaks were absent in the Ar-saturated solution (Fig. S18, ESI†). So, the overall mechanistic pathway of 4 e− mediated reduction of O2 to water schematically represented in Fig. S19, ESI.† To gain insight into the mechanism of O2 binding, we also performed computational studies on the weak interactions between covalent organic frameworks (COFs) and O2. The energy gaps of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were calculated for all the COP catalysts based on the molecular orbital (MO) diagrams. The HOMO–LUMO gaps were found to be 1.413 eV (RT-COP) and 1.529 eV (PT-COP) for end-on adsorption (Fig. 3(b) and (c)), and 1.450 eV and 1.566 eV for RT-COP and PT-COP, respectively, for side-on adsorption of the O2 molecule (Fig. S20a and b, ESI†). This indicates that the lower gap for RT-COP significantly enhances the interaction of the precursors via the Pauling model at the active sites during the ORR. The free energy of complexation for O2 with the COP materials was determined to be 16.2 kJ mol−1 for RT-COP and 12.7 kJ mol−1 for PT-COP, suggesting that the binding of O2 with RT-COP is more favourable than that with PT-COP. This weak interaction is consistent with the typical end-on adsorption (Pauling model) involving a carbon atom with Cδ+ (natural charges of 0.17 for RT-COP and 0.15 for PT-COP, as observed in natural bond orbital (NBO) calculations) (Fig. 3(d) and (e)), which is validated by previous studies.22,23 After adsorption of O2, the O–O bond distance was also elongated and found to be 1.251 & 1.245 Å in RT-COP and PT-COP, respectively (Fig. S22a and b, ESI†). Even the Wiberg bond index (WBI) in the case of RT-COF is lower than PT-COF (1.27 and 1.29 for O–O bond for RT-COF and PT-COF, respectively). All these observations provide strong evidence for the high ORR activity of RT-COP compared to the PT-COP catalyst material via the Pauling model. The strategically designed RT-COP polymer, with its more abundant keto groups, exhibits significant positive charges on the enamine carbon, which facilitates the electrochemical ORR through push–pull interactions.
 | ||
| Fig. 3 (a) In situ FTIR spectra of the RT-COP material in O2-saturated electrolyte solution. HOMO–LUMO gap of the 1:1 complex of (b) RT-COP, and (c) PT-COP catalysts. (d) Charge distribution curve of the RT-COP catalyst at 0.0004 iso-surface value on atoms. (e) Charge distribution curve of the PT-COP catalyst at 0.0004 iso-surface value on atoms. | ||
In summary, two extended π-conjugated covalent organic polymer framework materials with dual active sites have been synthesized. The RT-COP material demonstrates a moderately high half-wave potential, along with high durability and excellent methanol tolerance in alkaline media. Further experimental results are supported by density functional theory (DFT) analysis, which reveals the weak non-covalent push-and-pull interactions between adsorbed O2 at the catalytic active site, as well as the role of β-keto groups on adjacent carbon atoms in generating positive charges that enhance O2 adsorption. Strategic modulation of β-ketoenamine functionality along with the electron rich triazole unit synergistically effected the ORR mechanistic pathway. So, catalyst structure and activity were thoroughly demonstrated. This study emphasizes the importance of strategic design and synthesis of metal-free covalent organic polymers for achieving efficient electrochemical ORR processes.
SS thanks INST, Mohali, GK thanks UGC, New Delhi and TRKR thanks PMRF for the fellowship. RSD acknowledges BRNS (58/14/19/2023-BRNS) and DST SERB (CRG/2020/005683) for financial support.
Data availability
The data supporting this article are included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- D. W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576–591 RSC.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- X. Huang, et al. , Chem. Commun., 2022, 58, 8884–8899 RSC.
- S. Guo and S. Sun, J. Am. Chem. Soc., 2012, 134, 2492–2495 CrossRef CAS PubMed.
- X. Ge, et al. , ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- Y. Su, et al. , J. Am. Chem. Soc., 2024, 146, 15479–15487 CrossRef CAS PubMed.
- S. Wang, et al. , Nat. Commun., 2023, 14, 1–7 CAS.
- X. Chi, et al. , Angew. Chem., Int. Ed., 2024, e202418895 Search PubMed.
- H. Wang, et al. , Angew. Chem., Int. Ed., 2022, 61, e202202328 CrossRef CAS PubMed.
- L. Dai, et al. , Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- Z. Xiang, et al. , Polym. Chem., 2015, 6, 1896–1911 RSC.
- P. Peng, et al. , ACS Energy Lett., 2017, 2, 1308–1314 CrossRef CAS.
- D. Li, et al. , J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- K. Gong, et al. , Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- S. Haldar, et al. , J. Am. Chem. Soc., 2018, 140, 13367–13374 CrossRef CAS PubMed.
- Y. Hou, et al. , Environ. Sci. Technol., 2023, 57, 11675–11686 CrossRef CAS PubMed.
- C. R. Deblase, et al. , J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- T. Boruah, et al. , Chem. Commun., 2022, 58, 5506–5509 RSC.
- T. Murata, et al. , Commun. Chem., 2019, 2, 1–8 CrossRef CAS.
- R. Zhou, et al. , ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- G. Kumar, et al. , J. Mater. Chem. A, 2023, 11, 26508–26518 RSC.
- R. Ma, et al. , npj Comput. Mater., 2019, 5, 1–15 CrossRef CAS.
- Z. Shi, et al. , Electrochim. Acta, 2006, 51, 1905–1916 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, FESEM, XPS, additional electrochemical data regarding the ORR, and DFT calculations. See DOI: https://doi.org/10.1039/d4cc05271d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |