
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the effect of pressure on the crystal structure and caloric properties of the molecular ionic hybrid [(CH3)3NOH]2[CoCl4]†
Pedro
Dafonte-Rodríguez‡
 a,
Ignacio
Delgado-Ferreiro‡
a,
Javier
García-Ben
a,
Angel
Ferradanes-Martínez
a,
María
Gelpi
a,
Julian
Walker
b,
Charles James
McMonagle
c,
Socorro
Castro-García
a,
María Antonia
Señarís-Rodríguez
a,
Juan Manuel
Bermúdez-García
*a and
Manuel
Sánchez-Andújar
*a
a,
Ignacio
Delgado-Ferreiro‡
a,
Javier
García-Ben
a,
Angel
Ferradanes-Martínez
a,
María
Gelpi
a,
Julian
Walker
b,
Charles James
McMonagle
c,
Socorro
Castro-García
a,
María Antonia
Señarís-Rodríguez
a,
Juan Manuel
Bermúdez-García
*a and
Manuel
Sánchez-Andújar
*a
aUniversity of A Coruna, QuiMolMat Group, Dpt. Chemistry, Faculty of Science and Centro Interdisciplinar de Química e Bioloxía (CICA), Zapateira, 15071 A Coruña, Spain. E-mail: j.bermudez@udc.es; m.andujar@udc.es
bNorwegian University of Science and Technology, Department of Materials Science and Engineering, Trondheim 7491, Norway
cEuropean Synchrotron Radiation Facility, Swiss-Norwegian Beamlines, Grenoble 3843, France
First published on 6th November 2024
Abstract
The hybrid metal halide [(CH3)3NOH]2CoCl4 exhibits a first-order phase transition at T ∼ 343 K. Its crystal structure and caloric properties respond significantly to moderate pressures (1–1000 bar), demonstrating potential for applications in emerging solid-state refrigeration technologies.
Hybrid organic–inorganic metal halides have emerged as a versatile multifunctional material with a wide range of applications, including in the fields of photovoltaic energy,1 light-emitting diodes (LEDs),2 ferroelectricity,3 ferroelasticity,4 or piezoelectricity,5 among others. A common strategy for the development of new functional molecular materials is the construction of zero-dimensional (0D) hybrids combining discrete ionic components.6,7 These hybrid materials often involve the use of quasi-spherical organic cations and tetrahalometallate anions. The 0D nature of these materials allows for the incorporation of diverse components bonded together by weak intermolecular interactions. This facilitates solid–solid phase transitions and makes them well-suited for designing new multifunctional and multisensitive materials.8,9
In recent years, pressure-responsive phase change materials, such as hybrid perovskites,10,11 plastic crystals,12,13 polymers,14 or metal–organic frameworks15 among others have exhibited very large barocaloric effects (pressure-induced thermal changes in terms of isothermal entropy change ΔS, or adiabatic temperature change ΔT)16,17 of interest for gas-free eco-friendly refrigeration.
Nevertheless, only very few recent studies have reported barocaloric (BC) effects in hybrid metal-halide compounds.18,19
In this work, we focus on a new hybrid tetrahalometallate material [(CH3)3NOH]2[CoCl4], which is formed by the quasi-spherical N-hydroxyl ammonium cation [(CH3)3NOH]+ and the inorganic anion [CoCl4]2−. We conducted a comprehensive structural characterization of the phase transitions using variable-temperature and pressure synchrotron powder X-ray diffraction, as well as single-crystal X-ray diffraction. Additionally, we analysed the barocaloric response using two different methodologies: the quasi-direct method, which relies on isobaric differential scanning calorimetry (DSC) measurements under variable temperature, and the direct method, based on isothermal DSC under variable pressure.16,17 Furthermore, we established structure–property relationships to better understand the behaviour of the material.
This compound was synthesized via slow evaporation of aqueous solution containing the [(CH3)3NOH]+ cations and [CoCl4]2− anions (further details in ESI†). Dark blue powder and single crystals were obtained and confirmed to be the same single-phase by comparing experimental powder and single-crystal X-ray diffraction patterns (see Fig. S1 of ESI†).
Atmospheric pressure DSC experiments reveal that this compound undergoes a reversible and broad phase transition above room temperature at Th ∼ 343 K on heating and at Tc ∼ 338 K on cooling (Fig. S2 of ESI†). We obtain that the phase transition exhibits a relatively large latent heat of ΔH ∼ 33 kJ kg−1 and a thermally-induced entropy change of ΔS ∼ 105 J kg−1 K−1.
Single-crystal X-ray diffraction at different temperatures (T = 100 and 355 K) allowed us to determine the crystal structure above and below the observed phase transition temperature, which will be hereafter labelled as HT-phase (T > Tt) and RT-phase (T < Tt) for convenience.
The RT-phase shows a monoclinic symmetry with space group P21/n and lattice parameters a ∼ 8.5 Å, b ∼ 12.0 Å, c ∼ 15.0 Å and β ∼ 101° at T = 100 K (see Fig. 1a and Table S1 of ESI†).
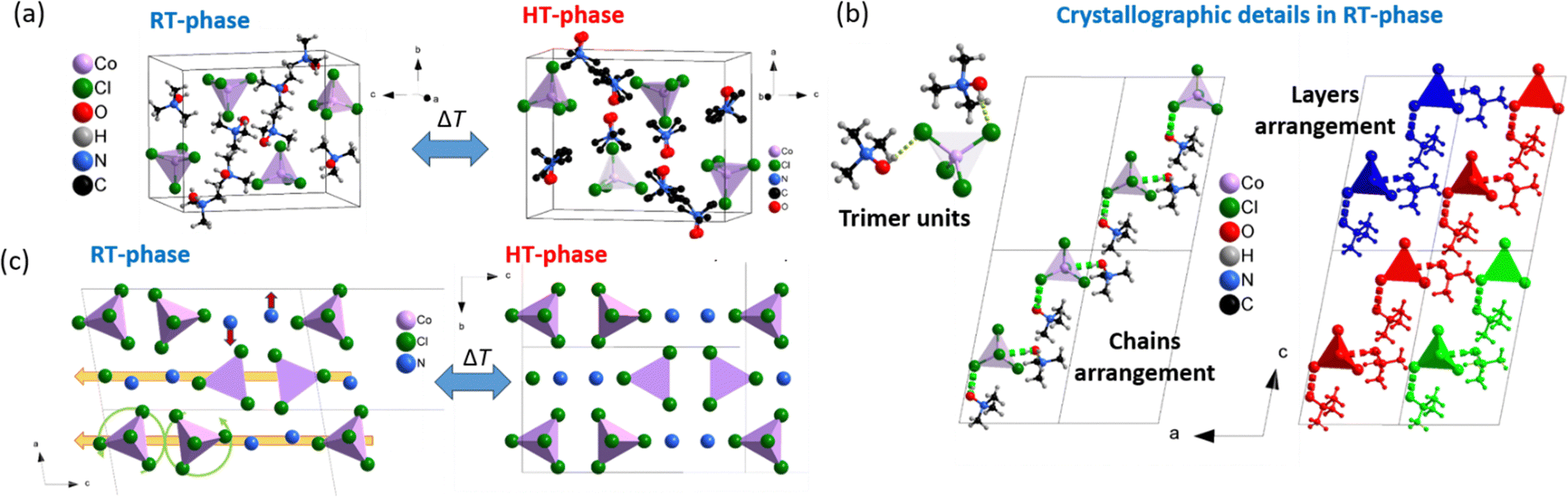 | ||
| Fig. 1 (a) Crystal structure of the RT- and HT-phase obtained by SCXRD. (b) Details of the RT-phase asymmetric unit forming trimers, and packing arrangement of trimers into chains, and chains into layers. (green dotted lines represent H-bonds). (c) Details of distortions present in the RT-phase in comparison with the HT-phase, namely, columnar shifts along the c-axis (yellow arrows), cooperative [CoCl4]− tetrahedra rotation (green curved arrows) and anti-parallel shift [(CH3)3NOH]+ cations (red arrows). Note: for simplicity, the [(CH3)3NOH]+ cations in Fig. 1c are depicted by their N-atoms only. | ||
The asymmetric unit consists of one [CoCl4]− anion and two [(CH3)3NOH]+ cations. The Co2+ cations adopt a slightly distorted tetrahedral geometry, with two Co–Cl bond lengths around 2.25 Å and the other two around 2.30 Å.
The Cl–Co–Cl bond angles range between 107.4 and 112.4 degrees, slightly deviating from the ideal angle of 109.5 degrees (further details in Table S2 and S3 of the ESI†).
Remarkably, the molecular interaction is distinguished by hydrogen bonding between the –OH groups of the organic cations and the Cl− anions. Each [CoCl4]− anion forms two hydrogen bonds with two different [(CH3)3NOH]+ cations forming an entity called a trimer, see Fig. 1b. It is worth noting that the observed distortion in geometry of [CoCl4]− anions is associated with these H-bonds.
On the other hand, the HT-phase crystallizes in an orthorhombic symmetry with space group Pnma and lattice parameters a ∼ 12.2 Å, b ∼ 8.7 Å and c ∼ 15.4 Å at T = 355 K (see Fig. 1 and Table S1 of ESI†). The main difference between both the RT- and HT-phases lies in an order–disorder process of the [(CH3)3NOH]+ cations. These cations are orientationally disordered in the HT-phase with the C and O atoms located in different crystallographic sites, while the [CoCl4]− anions remain ordered in both phases. Furthermore, several structural distortions were observed in the RT-phase compared to the HT-phase, specifically columnar shifts along the c-axis, which provoke β > 90°, a cooperative tilting of the [CoCl4]− tetrahedra, and an anti-parallel displacement of [(CH3)3NOH]+ cations along the a-axis (see Fig. 1c).
This phase transition can be classified as a ferroelastic transition according to Aizu notation (mmmF2/m),20 as previously reported for the analogous Zn compound.21
In order to explore the observed phase transition of this compound and to analyse its thermomechanical response and pressure responsiveness, we carried out an in situ study on variable temperature (VT) and pressure (VP) synchrotron powder X-ray diffraction (SPXRD) experiments.
From the analysis of the VT-SPXRD patterns (see Fig. S3 of ESI†), the phase transition can be clearly observed at T ∼ 330 K between RT- and HT-phase, which is consistent with our VT-DSC results. The obtained patterns were successfully refined using the Le Bail method, where the lattice parameters and space group were matched to those obtained by SCXRD.
According to these data, the lattice parameters exhibit a significant dependence on temperature (see Fig. S3 of the ESI†), with a singular behaviour that can be followed in 2 different temperature ranges: (1) upon heating from 260 to 290 K, all peaks shift to lower 2θ values, (2) when increasing the temperature up from 290 K to 330 K, some of these peaks still shift to even lower 2θ values, although some other peaks exhibit the opposite trend (see Table S4 of the ESI†). Overall, a conventional positive thermal expansion was observed for the low temperature range (T < 290 K). Meanwhile the RT-phase exhibits anomalous thermal expansion behaviour in the temperature range 290 to 330 K, in which there is a colossal TE in one direction a [201] = 406 (17) 10−6 K−1 together with negative expansion in the perpendicular one a [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 01] = −113 (7) 10−6 K−1. We propose that the anomalous thermal expansion observed is linked to the presence of the H-bonds and their specific packing arrangement. Notably, H-bonds are highly temperature-sensitive, weakening upon heating. As a result, the anomalous thermal expansion is most prominent just below the phase transition temperature, where these bonds experience significant weakening. The negative thermal expansion occurs along a specific direction corresponding to the chains formed by trimers and cooperative columnar displacement being the primary distortion responsible for this effect. Thus, we suggest that H-bonds are primarily responsible for the observed distortions in the RT-phase and the anomalous thermal expansion.
01] = −113 (7) 10−6 K−1. We propose that the anomalous thermal expansion observed is linked to the presence of the H-bonds and their specific packing arrangement. Notably, H-bonds are highly temperature-sensitive, weakening upon heating. As a result, the anomalous thermal expansion is most prominent just below the phase transition temperature, where these bonds experience significant weakening. The negative thermal expansion occurs along a specific direction corresponding to the chains formed by trimers and cooperative columnar displacement being the primary distortion responsible for this effect. Thus, we suggest that H-bonds are primarily responsible for the observed distortions in the RT-phase and the anomalous thermal expansion.
Additionally, in situ VP-SPXRD patterns were collected under isothermal conditions above and below the phase transition temperature and within the pressure range of 120–1000 bar using a bespoke sapphire capillary cell at the SNBL at ESRF.22
For T = 347 K, the phase transition is distinctly observed in the SPXRD patterns around 500 bar, indicating a transition from HT-phase to RT-phase with the pressure increase (see Fig. S4 of ESI†).
Moreover, we find a relatively large pressure region (between 300–600 bar) where both phases coexist. Furthermore, the VP-SPXRD patterns were fitted using the Le Bail method.
This allowed us to extract valuable information such as the variation of the lattice parameters and the beta angle in response to applied pressure (Fig. S4 of ESI†). In this context, we analysed the obtained data using the web-based tool PASCal v2 and we estimated the compressibility along the principal axes and the isothermal bulk modulus B0, using third-order Birch–Murnaghan equations. The calculated bulk modulus B0 was found to be 6.06 (3) GPa for the RT-phase at T = 325 K. This value is lower than in typical hybrid perovskites,23 and comparable to values found in metal–organic frameworks (MOFs).24 The compound exhibits an anisotropic compressibility, with the largest contraction occurring along the [201] direction and a smaller contraction (about half) along the [01] direction, coinciding with the chains formed by the trimers. Therefore, it seems that H-bonds add stiffness to the crystal structure, making it difficult for contraction along certain directions dominated by this intermolecular bonding.
Furthermore, as we observed by VT- and VP-DSC, these structural transitions and distortions with important contributions of H-bonds are responsible for significant barocaloric properties.
Our VT-DSC analysis at different pressures (quasi-direct methods) shows that the transition temperature shifted up to larger temperatures when increasing the pressure, which is indicative of a conventional barocaloric effect (dTt/dP > 0), see Fig. 2a and b.
 | ||
| Fig. 2 (a) |dQ/dT| versus T curves at different pressures, where peaks represent the heat flow of the phase transition. (b) T–p phase diagram built with the peak maximum observed by DSC on heating (red curve) and cooling (blue curve). (c) Volume change calculated by Le Bail fitting of the SPXRD patterns. (d) Barocaloric effects in term of isothermal entropy change ΔSit (coloured lines) and the reversible region ΔSrev (shaded areas) as obtained by quasi-direct methods. (e) Barocaloric effects in terms of reversible adiabatic temperature changes on applying (1 → p) and removing (p → 1) pressure. (f) Heat flow curves obtained by direct methods under isothermal conditions and under pressure ramps. | ||
From these data, we can obtain a pressure responsiveness (barocaloric coefficient) of (dTt/dp) ∼ 15.6 K kbar−1, which is similar to that of the best reported barocaloric hybrids.16,17
This barocaloric coefficient is in agreement to that estimated by the Clausius–Clapeyron equation (dTt/dp) = (ΔS/ΔV) ∼ 15.9 K kbar−1, where ΔS is the entropy change of the phase transition and ΔV is the volume change of the transition as obtained from SPXRD (Fig. 2c).
In addition, we calculated the barocaloric effects in terms of isothermal entropy change (ΔSit) by using quasi-direct methods, as reported elsewhere,16,17 see Fig. 2d:
| ΔSit = ΔSib (p ≠ 1,T) − ΔSib (p = 1,T) | (1) |
Since this compound exhibits a first-order phase transition with hysteresis under the application and removal of external stimuli, overcoming the transition hysteresis requires additional energy (pressure) that must be considered. Therefore, we determined the isothermally reversible entropy change (ΔSrev) that can be fully obtained under pressurization and depressurization cycles (see Fig. 2d). Interestingly, we found reversible barocaloric effects as large as ΔSrev ∼ 90 J K−1 kg−1 at T = 346 K under 1000 bar, a value which is similar or even slightly higher than those previously reported for related hybrid organic–inorganic plastic crystals, such as tetramethylammonium tetrachloroferrate [(CH3)4N][FeCl4] (ΔSrev ∼ 81 J K−1 kg−1 under Δp = 900 bar),18 quinuclidinium perrhenate [C7H14N][ReO4] (ΔSrev ∼ 60 J K−1 kg−1 under Δp = 1000 bar),18 [(CH3)3(CH2Cl)N][FeCl4] and [(CH3)3S][FeCl4] (ΔSrev ∼ 120 J K−1 kg−1 under p = 1000 bar).19
The calculated bulk moduli allowed us to estimate that the volumetric entropy change (ΔSv)25 is roughly one-third of the total entropy change, which suggests that the main contributions are related to additional factors, such as order–disorder processes and hydrogen bond cleavage and formation (see details in the ESI†).
In addition, we also calculated the adiabatic temperature change (ΔTrev) that can be reversibly obtained under pressure, which is as large as ΔTrev ∼ 8 K under 1000 bar, see Fig. 2e.
Additionally, we fully corroborate these results by performing direct calorimetric methods that are more precise although still scarce in the literature due to instrumental complexity. In this context, we carried out isothermal VP-DSC measurements during pressurization and depressurization cycles from 100–1000 bar with a rate of 10 bar min−1 and at different isothermal conditions of T = 346 K, 348 K and 350 K (Fig. 2f). The results reveal broad peaks associated with the phase transition induced by the applied pressure during both compression and decompression scans, which slightly decrease the observed entropy change with values of ΔSdirect ∼ 75 J K−1 kg−1. This observation is consistent with the in situ SPXRD patterns, which demonstrates that the pressure-driven phase transition occurs over a broad pressure range, where both phases coexist. It is noteworthy that within the studied pressure and temperature range, the phase transition exhibits full reversibility.
It is worth highlighting that the entropy value during the phase transition of this compound is comparable to that of other hybrid organic–inorganic plastic crystals, even if the latter experience severe orientational disorder. Meanwhile, the [(CH3)3NOH]2[CoCl4] compound exhibits less structural disorder during the phase transition, as only the organic cations become disordered while the inorganic anions remain ordered. We suggest that the presence of H-bonds between both organic cations and inorganic anions in [(CH3)3NOH]2[CoCl4] is partially responsible for the relatively large entropy values, despite showing a comparatively lower structural disorder. Even more, such characteristics facilitate the reversibility of the phase transition and enhance the long-term stability of the material, making it suitable for practical applications in future solid cooling devices. For further insights, we have performed similar studies on the [(CH3)3NOH]2[ZnCl4] compound, which reveal a similar barocaloric performance to the Co-compound (see Fig. S6 and Table S5 of ESI†).
Therefore, these findings will inspire the design and study of new barocaloric materials based on hybrid molecular compounds with this type of intermolecular bonding.
The authors area thankful for the financial support from grants PID2021-122532OB-I00 (MCIN/AEI & ERDF), PDC2021-121076-I00 (MCIN/AEI & EU-NG/PRTR), ED431C 2022/39 & ED431F 2023/33 (Xunta de Galicia), RYC2021-033040-I (MCIN/AEI & EU-NG/PRTR), UDC-Inditex InTalent and NTNU internal funding. The authors thank ESRF for the award of beam time (MA-5145, A01-2-1291 on BM01). Funding for open access publication granted by Universidade da Coruña/CISUG.
Data availability
The data supporting this article have been included as part of the ESI.† Further data will be available from authors upon reasonably request.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Kojima, et al. , J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- T. Hattori, et al. , Chem. Phys. Lett., 1996, 254, 103–108 CrossRef CAS.
- H. Zheng and K. P. Loh, Adv. Mater., 2024, 36, 1–18 Search PubMed.
- J. Li, et al. , Chem. – Eur. J., 2022, 28, e202201005 CrossRef CAS PubMed.
- V. Jella, et al. , Nano Energy, 2019, 57, 74–93 CrossRef CAS.
- M. Li and Z. Xia, Chem. Soc. Rev., 2021, 50, 2626–2662 RSC.
- S. Das, et al. , Chem. Soc. Rev., 2020, 49, 8878–8896 RSC.
- P. González-Izquierdo, et al. , J. Mater. Chem. C, 2021, 9, 4453–4465 RSC.
- J. Walker, et al. , Chem. Mater., 2022, 34, 2585–2598 CrossRef CAS.
- J. M. Bermúdez-García, et al. , Nat. Commun., 2017, 8, 15715 CrossRef.
- J. Seo, et al. , Nat. Commun., 2022, 13, 1–15 Search PubMed.
- B. Li, et al. , Nature, 2019, 567, 506–510 CrossRef CAS PubMed.
- P. Lloveras, et al. , Nat. Commun., 2019, 10, 1–7 CrossRef CAS.
- N. M. Bom, et al. , ACS Macro Lett., 2018, 7, 31–36 CrossRef CAS.
- M. Gelpi, et al. , Adv. Mater., 2023, 2310499, 1–9 Search PubMed.
- P. Lloveras and J.-L. Tamarit, MRS Energy Sustainable, 2021, 8, 3–15 Search PubMed.
- P. Lloveras, Barocaloric effects in the solid state: materials and methods, IOP Publishing, London, 2023 Search PubMed.
- A. Salvatori, et al. , J. Mater. Chem. A, 2023, 12140–12150 RSC.
- J. Salgado-Beceiro, et al., ChemRxiv, 2021, preprint, chemrxiv-2021-c4hx5 DOI:10.26434/chemrxiv-2021-c4hx5.
- K. Aizu, Phys. Rev. B: Solid State, 1970, 2, 754–772 CrossRef.
- W. Yuan, et al. , Chem. Commun., 2019, 55, 8983–8986 RSC.
- C. J. McMonagle, et al. , J. Appl. Crystallogr., 2020, 53, 1519–1523 CrossRef CAS.
- L. A. Muscarella, et al. , J. Phys. Chem. Lett., 2023, 14, 9042–9051 CrossRef CAS PubMed.
- J. C. Tan and A. K. Cheetham, Chem. Soc. Rev., 2011, 40, 1059–1080 RSC.
- A. Aznar, et al. , Appl. Mater. Today, 2021, 23, 101023 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2387156 and 2387157. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05125d |
| ‡ These authors have equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |