
Yolk-shelled MoS2/C@Void@C@MoS2 nanospheres as a stable and high-rate anode in sodium/potassium ion batteries†
Mang
Niu
*a,
Zongfan
Zhu
b,
Zhenkai
Mou
b and
Wenpei
Kang
 *b
*b
aState Key Laboratory of Bio-fibers and Eco-textiles, Institute of Biochemical Engineering College of Materials Science and Engineering, Qingdao University, Qingdao 266071, P. R. China. E-mail: mang.niu@qdu.edu.cn
bSchool of Materials Science and Engineering, China University of Petroleum (East China), Qingdao, 266580, P. R. China. E-mail: wpkang@upc.edu.cn
First published on 28th October 2024
Abstract
We report a yolk-shelled anode material with an MoS2/C core, large inner void and C@MoS2 shell (MoS2/C@Void@C@MoS2). This design could accelerate Na+/K+ reaction kinetics and endure volume changes. Based on the kinetics analysis and calculations based on density functional theory, this structure could effectively enhance the high-rate sodium/potassium storage capability.
Lithium-ion batteries have had higher requirements imposed on them owing to the rapid development of “clean” energy. However, the shortage of lithium resources has been the inherent bottleneck for their large-scale promotion. In recent years, sodium/potassium ion batteries (SIBs/PIBs) have been deemed to be promising novel cost-effective secondary battery candidates owing to rich resources and similar physicochemical properties.1,2 Nevertheless, the large-sized Na+/K+ will bring about serious volume effects and sluggish reaction kinetics, which limit the rate capability and worsen the lifespan of SIBs/PIBs. Therefore, it is meaningful to explore suitable electrodes with large channels for Na+/K+ transfer and rational structure to endure the volume changes.3
Fortunately, layered transition metal dichalcogenides (LTMDs) with large interlayers could facilitate Na+/K+ diffusion.4–6 As an essential representative of LTMDs, MoS2 with an interlayer spacing of 0.62 nm is regarded as an ideal candidate for Na+/K+ hosting, attributed to the weak interlayer van der Waals (vdW) force.7 Meanwhile, MoS2 anodes also suffer from spontaneous aggregation and unstable electrochemistry during the conversion reaction, which originate from the inherent poor conductivity and huge volume expansion. To address these issues, numerous efforts have been devoted to devise the architectures of LTMDs.8 Generally, a MoS2/carbon composite can increase the charge transfer and enhance its stability. On the other hand, nanostructure engineering can also decrease the diffusion distance of Na+, increase the interfacial charge storage, and effectively release the accumulated mechanical strain.4,9 Especially, as a unique type of hollow nanostructure, a yolk-shelled structure consisting of an active core and stable shell has large void spaces and rich interfacial boundaries, which provide sufficient spaces for volume expansion and enhance Na+/K+ reaction kinetics.10 Traditionally, yolk–shell structures are fabricated by several methods along the inside-to-outside and outside-to-inside directions. For example, the incorporation of iron in MoS2 (FMS@C) with a yolk–shell nanoarchitecture was prepared through a microemulsion strategy to deliver high structure stability, superior rate capability and impressive capacity retention.11 Besides, a few-layered MoS2@hollow porous carbon-sphere (MoS2@HPCS) with a yolk–shell structure was obtained in a hydrothermal process using a HPCS template, which boosted the K+ storage capability.12
Herein, a yolk shell-structured anode with a carbon coupled MoS2 core, carbon and MoS2 shell and large inner void (MoS2/C@Void@C@MoS2) was designed for SIBs and PIBs based on a hard-template strategy. This ingenious structure provides sufficient space for the volume expansion of the Na+/K+ insertion. Meanwhile, the shell with carbon and MoS2 can provide large channels for the Na+/K+ transfer and increase conductivity, and also strengthen the structure stability. As a result, the Na+/K+ storage capability can be boosted, achieving excellent specific capacities, rate performance, and cycling stability.
The preparation process of MoS2/C@Void@C@MoS2 is shown schematically in Fig. 1a. Through self-polymerization,13 the polypyrrole-phosphomolybdic acid (PPy-PMo12) precursor was obtained, which was coated by SiO2 and resorcinol–formaldehyde (RF) to create an inner void and carbon shell for the yolk–shell nanosphere. Through a carbonization and etching process, a hard template was obtained to grow MoS2 layers in a hydrothermal process, producing MoS2/C@Void@C@MoS2 with a yolk–shell structure. As shown in Fig. 1b, the X-ray diffraction (XRD) patterns were quite similar, related to the hexagonal MoS2 (JCPDS no. 37-1492). The peaks at 13.8°, 32.6°, 39.5° and 58.3° were related to the (002), (100), (103) and (110) planes of MoS2, respectively. Based on the peak position, the interplanar spacing of (002) planes was calculated to be 0.64 nm, which can provide large channels for the Na+/K+ transfer. It is obvious that the (002) peak became weak for MoS2/C@Void@C and MoS2/C@Void@C@MoS2, predicating the MoS2/C yolk to be well covered by the carbon shell. Raman spectra revealed the components of the samples (Fig. 1c). There were two strong bands at 378.5 cm−1 and 403.3 cm−1, belonging to the typical E12g (in-plane vibrational modes) and A1g modes (out-of-plane vibration) for the S–Mo bonds in MoS2,13,14 respectively. Another two bands at 1358.2 cm−1 (D band) and 1586.1 cm−1 (G band) were assigned to disordered carbon and vibration of sp2 carbon,15,16 respectively. The strong D band indicated the amorphous property of the carbon, providing more defect sites for the Na+/K+ reaction. The element oxidation states in MoS2/C@Void@C@MoS2 were also probed. We found that the involved Mo, S, C, N could be detected (Fig. S1, ESI†). The high-resolution Mo 3d spectrum (Fig. 1d) suggested that the peaks at 228.8, 232.1, 232.7 and 235.9 eV corresponded to Mo4+ 3d5/2, Mo4+ 3d3/2, Mo6+ 3d5/2 and Mo6+ 3d3/2, respectively, while another peak at 226.1 was assigned to S 2s.17 The existence of Mo6+ was ascribed to the surface oxidation of the MoS2.18 For S 2p, two distinct peaks at 161.6 eV (S 2p3/2) and 162.9 eV (S 2p1/2) confirmed the existence of S2− to form metal–S bonds. Another peak at 164.2 eV belonged to the S–C bond, demonstrating the strong interaction between MoS2 and carbon.19
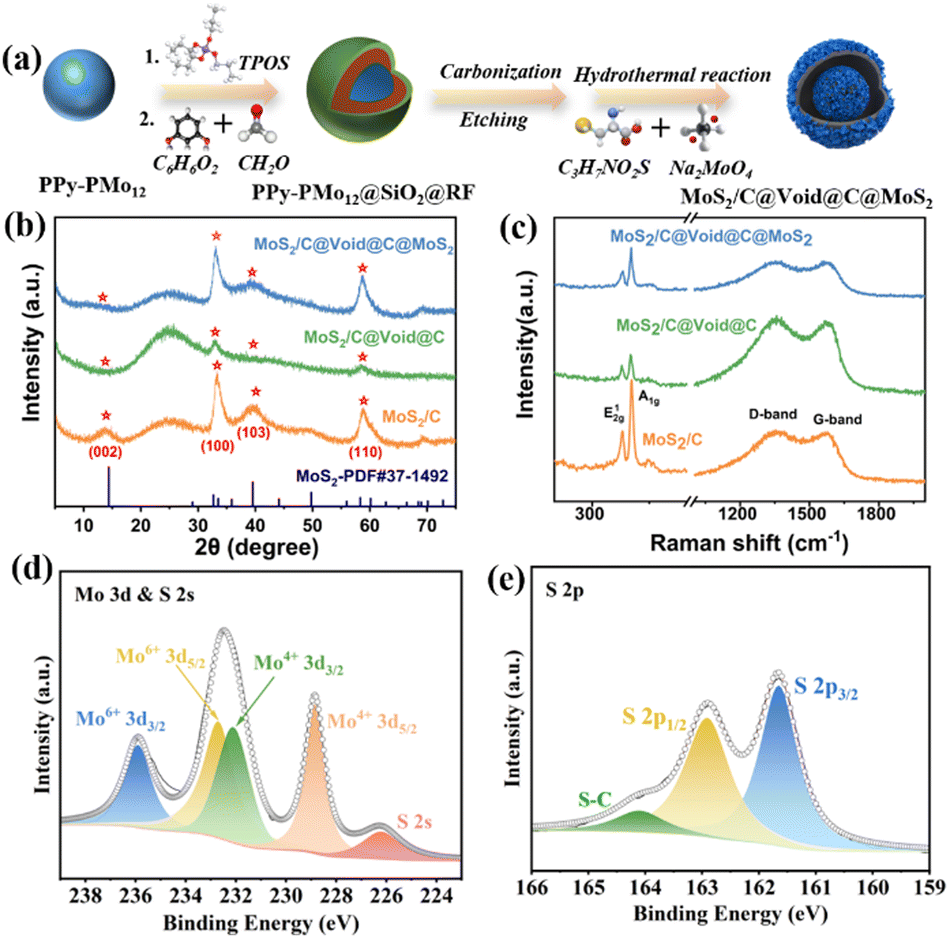 | ||
| Fig. 1 (a) Preparation of the yolk-shelled MoS2/C@Void@C@MoS2 nanosphere (schematic). (b) XRD patterns and (c) Raman spectra for the three samples. High-resolution XPS spectra of (d) Mo 3d and (e) S 2p. | ||
The structure was observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. 2). We found that PPy-PMo12 and PPy-PMo12@SiO2@RF were smooth nanospheres (Fig. S2a and b ESI†). The resultant MoS2/C nanosphere had numerous nanosheets on the surface (Fig. S2c, ESI†). For MoS2/C@Void@C (Fig. S2d, ESI†), smooth nanospheres were observed owing to the carbon shell construction. Fig. 2a shows the SEM image of MoS2/C@Void@C@MoS2 nanospheres, and the surfaces were well covered by nanosheets. The TEM image (Fig. 2b) showed a typical yolk–shell nanosphere comprising a solid core, large inner void and stable shell. High-resolution TEM images (Fig. 2c and d) showed that the nanosheets were tightly bonded with the carbon. The lattice fringe spacings of the nanosheets in both the yolk and shell were 0.64 nm, and assigned to the (002) plane of MoS2. The energy-dispersive spectroscopy (EDS) mapping images (Fig. 2e) demonstrated that Mo, S and C covered the whole yolk-shelled nanosphere, predicating the successful fabrication of MoS2/C@Void@C@MoS2.
 | ||
| Fig. 2 (a) SEM, (b) TEM and (c) and (d) HRTEM images, and (e) EDX elemental mapping images of Mo, S and C for the MoS2/C@Void@C@MoS2. | ||
Considering the superiority of the MoS2/C@Void@C@MoS2 structure, it was used as anode material for SIBs/PIBs. Fig. 3a shows the CV curves of SIBs. There were five peaks in the first cycle. The peaks at 1.2 V and 0.95 V corresponded to the Na+ intercalation into the core and shell MoS2 interlayers to form NaxMoS2.11,12 The peak at 0.6 V was ascribed to the formation of solid electrolyte interface (SEI) layers on enriched interfacial boundaries, which would lead to irreversible capacity loss and low initial coulombic efficiency (ICE).20 The peak at 0.38 V was assigned to the conversion reaction from NaxMoS2 to metallic Mo and NaxS.21 The strong peak at 0.12 V should have originated from Na+ storage in the electrode interfaces and carbon skeletons.22 The peaks for the subsequent four cathodic cycles moved to higher voltages owing to electrode activation. For the oxidation process, the weak peak at 0.37 V was assigned to Na+ removal from the interfaces and carbon.23 The peak at 1.8 V was attributed to the reversible oxidation reaction for form MoS2. Moreover, the CV curves almost overlapped from the second-to-fifth cycle, predicating its stable cycling performance. Galvanostatic charge/discharge cycling was measured. Fig. S3 (ESI†) shows selected discharge/charge profiles and the related dQ/dV curves. The difference of the first discharge profile was ascribed to SEI formation and electrode amorphization. Fig. 3b shows the rate performance, delivering capacities of 463.2, 433.3, 412.0, 393.2, 367.1 and 342.0 mA h g−1 at 0.2, 0.5, 1.0, 2.0, 5.0 and 10.0 A g−1, respectively. High capacity retention of 73.8% could be maintained when the current increased 50-fold. The superior rate capability should have originated from the ingenious yolk–shell structure and carbon coupling, which can decrease ion-transfer distances and increase the conductivity. The cycling performance of MoS2/C@Void@C@MoS2 was further evaluated and compared with that of MoS2/C and MoS2/C@Void@C at 5.0 A g−1. The first discharge/charge capacities with an activation current of 50 mA g−1 were 1203.8/672.5 mA h g−1. After that, the capacity was 402 mA h g−1 in the second cycle at 5.0 A g−1, and the capacities were stable in the initial 300 cycles. After 1000 cycles, the capacity was kept at 313 mA h g−1, giving a high capacity retention of 77.9%. In contrast, MoS2/C showed high capacity in the initial cycles, which had obvious decay during cycling. Also, 213 mA h g−1 was achieved after 1000 cycles, giving a capacity retention of 48.5%. MoS2/C@Void@C showed a low capacity of 240 mA h g−1 in the second cycle with high capacity retention of 77.8%, benefiting from the yolk–shell structure and carbon protection. Consequently, the carbon shell could stabilize the cycling performance, while the outer MoS2 layer could increase the reaction activity for Na+ storage. The MoS2/C@Void@C@MoS2//Na3V2(PO4)3@C full cell also showed impressive electrochemical performance (Fig. S5 ESI†).
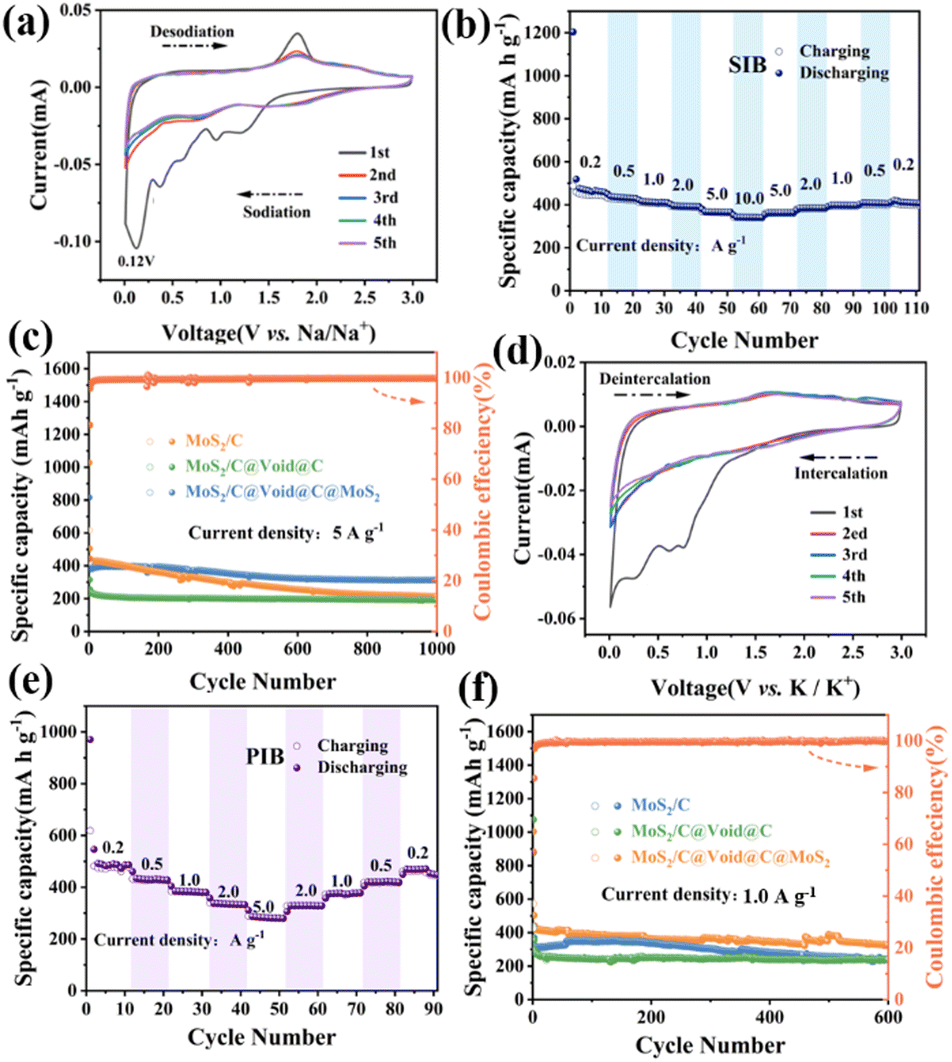 | ||
| Fig. 3 (a) CV curves at a scan rate of 0.1 mV s−1 and (b) rate performance of MoS2/C@Void@C@MoS2 in SIBs. (c) Cycling comparison at 5.0 A g−1 after activation with 50 mA g−1 in the first cycle for SIBs. (d) CV curves at 0.1 mV s−1 and (e) rate capability of MoS2/C@Void@C@MoS2 in PIBs. (f) Cycling comparison at 5.0 A g−1 after activation with 50 mA g−1 in the first cycle for PIBs. | ||
In view of the superiority of MoS2/C@Void@C@MoS2 in SIBs, it was further studied as anode material in PIBs. The first CV curve (Fig. 3d) showed three obvious peaks. The peaks above 0.5 V were assigned to K+ intercalation into MoSe2 layers to form KxMoSe2, accompanied by the formation of SEI layers.24,25 The peak at 0.26 V was related to the conversion from KxMoSe2 to metallic Mo.25 In the subsequent anodic process, the weak peak between 1.5 and 2.5 V was related to the reversible formation of MoSe2. The curves in the following cycles almost overlapped. The selected voltage profiles and dQ/dV curves are shown in Fig. S4 (ESI†), and gave similar results to the CV. The rate performance was measured at 0.2, 0.5, 1.0, 2.0 and 5.0 A g−1, delivering capacities of 486.1, 430.3, 385.4, 336.5 and 287.1 mA h g−1, respectively (Fig. 3e). When the current returned to 0.2 A g−1, the capacity could recover almost to the initial capacity of 470.5 mA h g−1, revealing a good rate capability. Meanwhile, its cycling performance was compared with that of MoS2/C and MoS2/C@Void@C at 1.0 A g−1 (Fig. 3f). MoS2/C@Void@C@MoS2 delivered first discharge/charge capacities of 1003.4/573.1 mA h g−1 with an activated current of 50 mA g−1, producing an ICE of 57.1%. MoS2/C@Void@C@MoS2 showed higher capacities than those for the MoS2/C and MoS2/C@Void@C over the whole cycling at 1.0 A g−1. It showed capacity of 503.5 mA h g−1 in the second cycle and 326.9 mA h g−1 after 600 cycles, which were higher than those for MoS2/C (356.0 mA h g−1 and 239.7 mA h g−1) and MoS2/C@Void@C (369.2 mA h g−1 and 239.7 mA h g−1). These data demonstrated that MoS2/C@Void@C@MoS2 also possessed superior cycling and rate performance in PIBs owing to the synergistic effect of the carbon@MoSe2 shell and large voids in the yolk–shell structure.
To explore the underlying promotion of the high-rate Na+/K+ storage capability, kinetic properties were performed through capacity contribution analysis, and comparison of electrochemical impedance spectroscopy (EIS), galvanostatic intermittent titration technique (GITT) measurements and density functional theory (DFT) calculations. Attributed to enriched interfacial boundaries, the charge storage was contributed by both faradaic and pseudocapacitive processes.26 According to the analysis of CV curves with different scan rates of the MoS2/C@Void@C@MoS2 anode, the capacity contribution could be separated (Fig. S6, ESI†).27,28 The capacitive contribution gradually increased along with an increase in the scan rate, and could reach 93% at 1.0 mV s−1 from 82% at 0.2 mV s−1. These data revealed the high-rate sodium capability to be mainly promoted by the capacitive behaviour. Similarly, the superior high-rate K+-storage capability was also dominated by the capacitive contribution (Fig. S7, ESI†). Moreover, the enhanced reaction kinetics of MoS2/C@Void@C@MoS2 were confirmed by EIS and GITT. Fig. 4a and b show the Nyquist plots for SIBs and PIBs, which had similar shapes. The charge-transfer resistance (Rct) of the MoS2/C@Void@C@MoS2 anode in the semicircle region was the lowest among the three electro des both in SIBs and PIBs, implying its higher ionic conductivity. Solid-state diffusion of Na+/K+ (DNa/DK) also affected the reaction kinetics, and the GITT could be used to determine the DNa and DK values in the whole charging/discharging process.29 Based on Fick's second law,30 it became obvious that MoS2/C@Void@C@MoS2 had higher DNa and DK values (Fig. S8, ESI†). These data suggested that the yolk–shell structure with a C@MoS2 shell could boost the reaction kinetics for Na+/K+. DFT calculations were further performed to reveal the diffusion energy barrier (Eb) of Na+ and K+ across C and C@MoS2 shells. The climbing image nudged elastic band (CI-NEB) method was used to calculate the diffusion path and Eb of Na+ and K+.31,32 The optimized Na+ and K+ diffusion paths are shown in Fig. S9 (ESI†). Obviously, the Eb of 0.034 eV for Na+ and 0.013 eV for K+ across the C@MoS2 shell was lower than those for the C shell. This finding suggested that introduction of MoS2 on the carbon shell could reduce the Eb and, thus, facilitate the diffusion of Na+ and K+, resulting into improved reaction kinetics and activity.
 | ||
| Fig. 4 Nyquist plots of MoS2/C, MoS2/C@Void@C and MoS2/C@Void@C@MoS2 anodes for (a) SIBs and (b) PIBs. Diffusion barriers of (c) Na+ and (d) K+ across carbon and the carbon@MoS2 shell. | ||
In summary, yolk-shelled MoS2/C@Void@C@MoS2 was synthesized and demonstrated to be superior anode material for SIBs and PIBs. This interesting structure could effectively buffer volume changes, facilitate Na+/K+ diffusion and fast charge transfer. These features could increase conductivity and DNa/DK values, and decrease the Eb of Na+/K+, achieving superior high-rate performance in SIBs/PIBs. This work illustrates the importance of the yolk–shell structure and shell design, opening up a way to construct advanced anode materials for SIBs/PIBs.
Mang Niu: methodology, investigation, and writing (original draft, review and editing). Zongfan Zhu: methodology and investigation. Zhenkai Mou: methodology and investigation. Wenpei Kang: supervision, conceptualization, writing (review and editing), project administration, and funding acquisition.
This work was financially supported by the National Natural Science Foundation of China (52474336) and the Natural Science Foundation of Shandong Province (ZR2022MB084).
Data availability
The data supporting the results of this study have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- F. Huang, P. Xu, G. Fang and S. Liang, Adv. Mater., 2024, 2405310 CrossRef CAS.
- Z. He, Y. Huang, G. Zou, H. Liu, H. Hou, Z. Geng and X. Ji, Nano Energy, 2024, 129, 109996 CrossRef.
- Y. Wang, W. Kang and D. Sun, ChemSusChem, 2023, 16, e202202332 CrossRef PubMed.
- J. Rehman, S. Liu, M. K. Butt, X. Fan, T. Khattab, K. A. Elsayed and M. F. Shibl, Chem. Eng. J., 2023, 461, 141924 CrossRef.
- J. Xu, J. Zhang, W. Zhang and C. Lee, Adv. Energy Mater., 2017, 7, 1700571 CrossRef.
- M. Ma, Y. Yao, Y. Wu and Y. Yan, Adv. Fiber Mater., 2020, 2, 314 CrossRef.
- J. Li, Y. Zhang, Y. Mao, Y. Zhao, D. Kan, K. Zhu, S. Chou, X. Zhang, C. Zhu, J. Ren and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e20230305 Search PubMed.
- J. Wu, F. Ciucci and J.-K. Kim, Chem. – Eur. J., 2020, 26, 6296 CrossRef PubMed.
- B. Chen, D. Chao, E. Liu, M. Jaroniec, N. Zhao and S.-Z. Qiao, Energy Environ. Sci., 2020, 13, 1096 Search PubMed.
- C. Qin, Z.-J. Jiang, T. Maiyalagan and Z. Jiang, Chem. Rec., 2024, 24, e202300206 CrossRef CAS PubMed.
- M. Chu, X. Xu, W. Zhao, Y. Dai, X. Zhou, G. Xue, Y. Xue, L. Cao and S. Chen, ACS Sustainable Chem. Eng., 2024, 12, 12596 CrossRef CAS.
- J. Hu, Y. Xie, X. Zhou and Z. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 1232 CrossRef CAS.
- X. Wang, W. Kang, Y. Wang, B. Zhang and D. Sun, J. Mater. Chem. A, 2021, 9, 17780 RSC.
- H. Xia, L. Zan, P. Yuan, G. Qu, H. Dong, Y. Wei, Y. Yu, Z. Wei, W. Yan, J. S. Hu, D. Deng and J. N. Zhang, Angew. Chem., Int. Ed., 2023, 62, 202218282 CrossRef.
- X. Li, R. Wang, Q. Wu, Y. Yu, T. Gao, T. Yao, X. Wang, J. Han and B. Song, Small, 2023, 19, 2206940 CrossRef CAS PubMed.
- N. Nagmani, S. Manna and S. Puravankara, Chem. Commun., 2024, 60, 3071 RSC.
- G. K. Veerasubramani, M. S. Park, H. S. Woo, Y. K. Sun and D. W. Kim, Energy Storage Mater., 2021, 38, 344 CrossRef.
- T. Wang, K. Yao, Y. Hua, E. G. Shankar, R. Shanthappa and J. S. Yu, Chem. Eng. J., 2023, 457, 141363 CrossRef CAS.
- Y. Jiao, A. Mukhopadhyay, Y. Ma, L. Yang, A. M. Hafez and H. Zhu, Adv. Energy Mater., 2018, 8, 1702779 CrossRef.
- J. Ru, T. He, B. Chen, Y. Feng, L. Zu, Z. Wang, Q. Zhang, T. Hao, R. Meng, R. Che, C. Zhang and J. Yang, Angew. Chem., Int. Ed., 2020, 59, 14621 CrossRef CAS PubMed.
- Y. Zhou, Y. Liu, M. Zhang, Q. Han, Y. Wang, X. Sun, X. Zhang, C. Dong, J. Sun, Z. Tang and F. Jiang, Chem. Eng. J., 2022, 433, 133778 CrossRef CAS.
- L. Shi, Y. Sun, W. Liu, F. Zhao, R. Liu, C. Dong, G. Cheng and J. Ding, Chem. Commun., 2023, 59, 12723 RSC.
- X. Chen, J. Tian, P. Li, Y. Fang, Y. Fang, X. Liang, J. Feng, J. Dong, X. Ai, H. Yang and Y. Cao, Adv. Eng. Mater., 2022, 12, 2200886 CAS.
- T. Yang, J. Liang, I. Sultana, M. M. Rahman, M. J. Monteiro, Y. Chen, Z. Shao, S. R. P. Silva and J. Liu, J. Mater. Chem. A, 2018, 6, 8280 RSC.
- Y. Cui, W. Liu, W. Feng, Y. Zhang, Y. Du, S. Liu, H. Wang, M. Chen and J. Zhou, Adv. Funct. Mater., 2020, 30, 1908755 CrossRef CAS.
- Y. Gao, P. Hai, L. Liu, J. Yin, Z. Gan, W. Ai, C. Wu, Y. Cheng and X. Xu, ACS Nano, 2022, 16, 14745 CrossRef CAS PubMed.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925 CrossRef CAS.
- X. Xu, J. Liu, J. Liu, L. Ouyang, R. Hu, H. Wang, L. Yang and M. Zhu, Adv. Funct. Mater., 2018, 28, 1707573 CrossRef.
- X. Rui, N. Yesibolati, S. Li, C. Yuan and C. Chen, Solid State Ion., 2011, 187, 58 CrossRef CAS.
- C. An, Y. Yuan, B. Zhang, L. Tang, B. Xiao, Z. He, J. Zheng and J. Lu, Adv. Energy Mater., 2019, 9, 1900356 CrossRef.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978 CrossRef.
- D. Sheppard, R. Terrell and G. Henkelman, J. Chem. Phys., 2008, 128, 134106 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05088f |
| This journal is © The Royal Society of Chemistry 2024 |