
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of phosphate stabilised iodanes and their application in intramolecular aryl migrations†
Jan Rick
Koch
 a,
Mattis
Damrath
a,
Pim
Puylaert
b and
Boris J.
Nachtsheim
*a
a,
Mattis
Damrath
a,
Pim
Puylaert
b and
Boris J.
Nachtsheim
*a
aInstitute for Organic and Analytical Chemistry, University of Bremen, Leobener Straße 7, 28359 Bremen, Germany. E-mail: nachtsheim@uni-bremen.de
bInstitute for Inorganic Chemistry and Crystallography, University of Bremen, Leobener Straße 7, 28359 Bremen, Germany
First published on 6th November 2024
Abstract
A diverse set of hydroxy-benzo[e]iodadioxaphosphinine oxides and derived diaryl iodonium salts are prepared and two examples are characterized by X-ray crystallography, featuring an out-of-plane geometry of the hypervalent bond for both compound classes. Treatment of the phosphate-stabilized diaryliodonium salts with Ca(OH)2 results in an efficient base-induced intramolecular aryl migration under aqueous conditions, yielding iodo-substituted diaryl ethers with yields up to 94%. Our findings highlight the synthetic potential of this previously underexplored compound class in organic transformations.
Hypervalent iodine compounds are versatile reagents for oxidative transformations in organic synthesis.1 Substrates like phenyliodine(III)diacetate (PIDA), 2-iodoxybenzoic acid (IBX) and Dess–Martin–Periodinan (1, DMP) can be used in alcohol oxidations,2 alkene functionalization3 or heterocycle syntheses.4 Most O-stabilized iodanes like IBX or DMP are characterized by a cyclic benziodoxolone structure, leading to higher thermal stability and more controlled reactivity.5 Such beneficial cyclic structures can also be accomplished with alcohol functionalities6 (2) or through pseudo-cyclic interactions with tethered N-heterocycles7 (3) or sulfonyl-groups8 (4, Fig. 1A). So far, only a limited amount of phosphorus oxoacid substituted iodanes were described. Examples include [hydroxy(phosphoryloxy)iodo]arenes 5 and its cyclic derivative 6. While most phosphorus-containing iodanes like 5 and 6 are based on P–O ligands at the I(III) center,9 only a few examples of intramolecular P–O-stabilization are known. Balthazor et al. reported the synthesis of the five-membered iodane 7 (Fig. 1B), whereas Protasiewicz et al. published the synthesis of a phosphorus-containing iodane with a pseudo cyclic structure.10 The first intramolecular phosphate stabilized iodane (8a) was synthesized by J. E. Leffler and H. Jaffe in 1973 by treating the corresponding phosphoric acid with peracetic acid.11 However, these initial investigations are focused on the structure of these compounds without investigating their further reactivity. Herein, we want to present the synthesis of various phosphate-substituted iodanes, their transformation into the stabilized-diaryl derivatives 9 and their reactivity in a base induced aryl migration (10, Fig. 1C).
 | ||
| Fig. 1 (A) Common intramolecular stabilized iodanes 1–4, (B) P–O-stabilized iodanes 5–8, (C) aim of this work. | ||
Starting with the oxidation of various o-iodo phenyl phosphates (11), the corresponding cyclic iodosophenylphosphoric acids 8a–f and 8h–i were obtained by using mCPBA as the oxidant (Scheme 1). The established oxidation reagents like Oxone® and Selectfluor® were incompatible due to the formation of various side products like multiple fluorinations. Electron-rich and poor substituted iodanes (8a–f, 8h–i) in ortho-, meta- and para-position regarding the iodine center was obtained in high yields of up to 99%. The acetylated derivate 8g was not accessible due to incomplete oxidation. The 4- and 5-methoxy derivatives could be synthesized, but they were obtained as impure materials and proved to be too unstable for further transformations. Substrates with a second ortho-substituent next to the iodine (8j–k) could also not be isolated due to the ortho-effect in stabilised-iodanes.12 Also, instead of the desired oxidation instant deiodination was observed. We obtained the X-ray structure of the methoxy ligand-exchanged derivate 8aOMe and gained further insights into the solid-state structure. Therein, the cyclic structure was confirmed with an I1–O1 distance of 2.27 Å and an O1–I1–C1–C2 angle of 43.8° which generates an out-of-plane geometry.
 | ||
Scheme 1 Oxidation of o-iodo phenyl phosphates into iodosophenylphosphoric acids 8a–k and X-ray structure of 8aOMe. General reaction conditions: 11 (1.00 eq.), mCPBA (2.00 eq.), DCM (0.2 M), stirring 5 min at rt and 2 h at 60 °C. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) reaction at rt with 1.20 eq. mCPBA. reaction at rt with 1.20 eq. mCPBA. | ||
We then further examined their reactivity and tried to synthesize their diarylated analogues (Scheme 2). By using an excess of mesitylene, as suitable electron-rich arene for the coupling, in an MeCN/TFA mixture, we were able to generate the ortho-phosphate stabilized species 9a–h in high yields of up to 94%. The synthesis of other strong electron-rich derivatives 9i–j and 9n can be carried out in high yields of up to 92% but reaches its limits already for tert-butylbenzene. The regioselective coupling of arenes with more than one reactive side was achieved using an excess of their trimethylsilyl substituted derivatives in MeCN/TFA. Using this method, we were able to obtain the less electron rich compounds 9k–m, 9o–p and 9r in yields from 65–83%. Utilization of one equivalent of the fluorinated and chlorinated trimethylsilyl arenes suppressed the formation of an undesired diaryliodonium species. Arenes with stronger electron-withdrawing trifluoromethyl groups could not be converted into the desired product 9q. Again, we were able to obtain a crystal structure for the phosphate-stabilized iodane 9a, which shows the same cyclic geometry, but with an elongated I1–O1 bond length of 2.99 Å whereas the O1–I1–C1–C2 angle of 40.3° creates a similar out-of-plane geometry as observed in 8a.
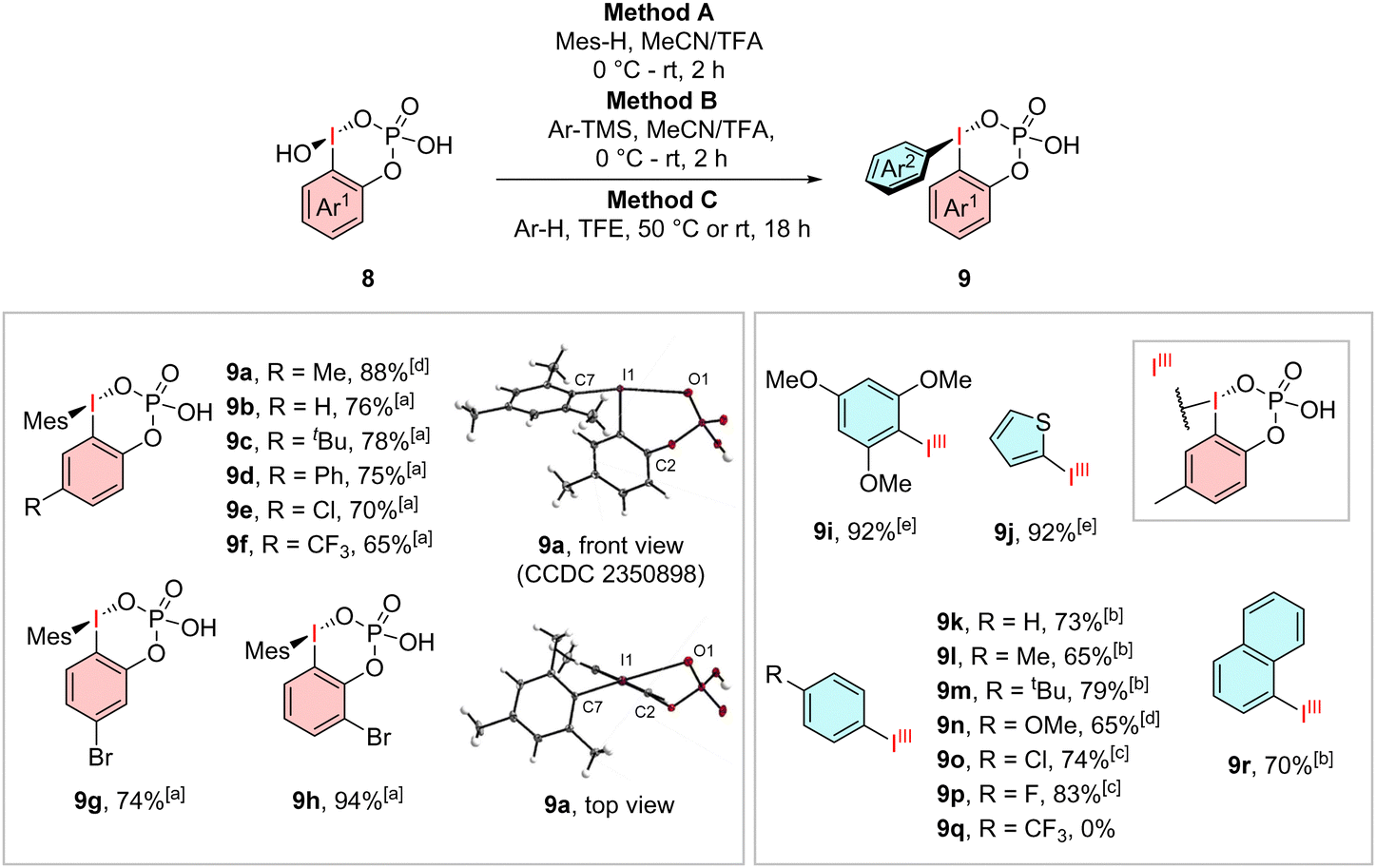 | ||
| Scheme 2 Arylation of iodosophenylphosphoric acids into 9a–r and X-ray structure of 9a. General reaction conditions: amethod (A) 8 (1.00 eq.), mesitylene (2.00 eq.), MeCN/TFA (1:1, 0.1 M), stirring 5 min at 0 °C and 2 h at rt. bMethod B: 8 (1.00 eq.), TMS-arene (1.50 eq.), MeCN/TFA (1:1, 0.1 M), stirring 5 min at 0 °C and 2 h at rt. cMethod B with 1.00 eq. of TMS-arene. dMethod C: 8 (1.00 eq.) arene (2.00 eq.), TFE (0.1 M), stirring at 50 °C for 18 h. eMethod C with stirring at rt. | ||
To demonstrate the reactivity of these phosphate-stabilized diaryliodonium salts the aryl migration into diaryl ethers was investigated. A reaction which was previously described for ortho-triflate-substituted iodonium salts by Han et al.13 Starting with the literature conditions of Cs2CO3 in MeCN the diaryl ether 10a was obtained in a high yield of 98%, whereas the reaction was carried out at 80 °C instead of room temperature. Due to their potential physiological relevance, we investigated this reaction under aqueous conditions (see ESI,† Table S1) and obtained the diaryl ether 10a in a yield of 81% with the use of Ca(OH)2 as base at 40 °C. Under these optimized conditions, all the previously synthesized arylated iodanes 9 were examined (Scheme 3). Different substitutions in meta- and para-position to the iodine center caused a minor variance in the reactivity, which yielded the ethers 10b–e and 10g in up to 79%. The CF3-substituted derivative 10f was isolated in 51% yield. Another substitution in the second meta-position to the iodine also resulted in diminished yields of 51% as observed in product 10h. The phenyl-substituted derivative 10i yielded the diarylether in 91%, whereas the toluyl-substituted compound 10j was obtained in a lower yield of 65%. Even more electron-rich arenes, like tert-butyl- and methoxy-substituted derivates 10k and 10l, resulted in moderate yields of 55% and a low yield of 14%, respectively. For the latter the yield could be increased to 22% by conducting the reaction at room temperature.
 | ||
| Scheme 3 Substrate scope of diaryl ethers 10a–q synthesized from 9. General reaction conditions: 9 (100 μmol), Ca(OH)2 (2.00 eq.), H2O (2 mL), 40 °C, 24 h. aAt room temperature, b48 h reaction time. | ||
In contrast to the high yield of 94% for the chlorinated derivative 10m, the fluorinated ether 10n was obtained just in a moderate yield of 52%, while the main side product was generally the iodinated arene as mentioned in the optimization (see ESI,† Table S1). Furthermore, the TMP-, naphthalene- and thiophene compounds 10o, 10p, and 10q were obtained in good yields of up to 70%.
In summary, we synthesized a variety of different iodosophenylphosphoric acids, which could be subsequently converted towards their phosphate-stabilized diarylated species through ligand exchange with different arenes. The X-ray data of both compounds confirmed a cyclic structure that leads to an out-of-plane distortion of the hypervalent bond. These novel arylated iodanes could successfully applied in base-induced intramolecular aryl migration in an aqueous medium to obtain various iodo-substituted diaryl ethers in yields up to 94%. Since this interesting reactivity was observed in aqueous solution under ambient conditions a potential physiological relevance might be operational and is part of further investigations in our laboratory.
Data availability
The data supporting this study, including detailed experimental procedures, spectroscopic data, and additional information, are available in the ESI.† Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers: 2350899 and 2350898.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (b) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed; (c) C. Rocq, M. Denis and S. Canesi, Chem. Commun., 2023, 59, 6495–6508 RSC; (d) F. V. Singh, S. E. Shetgaonkar, M. Krishnan and T. Wirth, Chem. Soc. Rev., 2022, 51, 8102–8139 RSC; (e) X. Peng, A. Rahim, W. Peng, F. Jiang, Z. Gu and S. Wen, Chem. Rev., 2023, 123, 1364–1416 CrossRef CAS PubMed.
- (a) J. Yadav, B. Reddy, A. Basak and A. Venkat Narsaiah, Tetrahedron, 2004, 60, 2131–2135 CrossRef CAS; (b) M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086–2099 RSC; (c) K. Bensberg, A. Savvidis, F. Ballaschk, A. Gómez-Suárez and S. F. Kirsch, Chem. – Eur. J., 2024, 30, e202304011 CrossRef CAS PubMed.
- (a) L. Emmanuvel, T. M. A. Shaikh and A. Sudalai, Org. Lett., 2005, 7, 5071–5074 CrossRef CAS PubMed; (b) T. Kitamura, K. Yoshida, S. Mizuno, A. Miyake and J. Oyamada, J. Org. Chem., 2018, 83, 14853–14860 CrossRef CAS PubMed.
- (a) L.-H. Du and Y.-G. Wang, Synthesis, 2007, 675–678 CAS; (b) H. Kawai, S. Okusu, E. Tokunaga and N. Shibata, Eur. J. Org. Chem., 2013, 6506–6509 CrossRef CAS.
- (a) A. Yoshimura, A. Saito and V. V. Zhdankin, Adv. Synth. Catal., 2023, 365, 2653–2675 CrossRef CAS; (b) A. Boelke, Y. A. Vlasenko, M. S. Yusubov, B. J. Nachtsheim and P. S. Postnikov, Beilstein J. Org. Chem., 2019, 15, 2311–2318 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- (a) A. Boelke, E. Lork and B. J. Nachtsheim, Chem. – Eur. J., 2018, 24, 18653–18657 CrossRef CAS PubMed; (b) A. Boelke, S. Sadat, E. Lork and B. J. Nachtsheim, Chem. Commun., 2021, 57, 7434–7437 RSC.
- D. Macikenas, E. Skrzypczak-Jankun and J. D. Protasiewicz, Angew. Chem., Int. Ed., 2000, 39, 2007–2010 CrossRef CAS PubMed.
- (a) R. A. Moss and H. Zhang, J. Am. Chem. Soc., 1994, 116, 4471–4472 CrossRef CAS; (b) G. F. Koser, J. S. Lodaya, D. G. Ray and P. B. Kokil, J. Am. Chem. Soc., 1988, 110, 2987–2988 CrossRef CAS.
- (a) B. V. Meprathu, M. W. Justik and J. D. Protasiewicz, Tetrahedron Lett., 2005, 46, 5187–5190 CrossRef CAS; (b) T. M. Balthazor, J. A. Miles and B. R. Stults, J. Org. Chem., 1978, 43, 4538–4540 CrossRef CAS.
- J. E. Leffler and H. Jeffe, J. Org. Chem., 1973, 38, 2719–2721 CrossRef CAS.
- K. N. Parida and J. N. Moorthy, Chem. – Eur. J., 2023, 29, e202203997 CrossRef CAS PubMed.
- H. Chen, J. Han and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 12313–12317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data (1H-, 13C- and 19F-NMR-chemical shifts, IR-bands, melting points) including the corresponding NMR-spectra and X-ray data can be found in the supporting information. CCDC 2350898 and 2350899. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04934a |
| This journal is © The Royal Society of Chemistry 2024 |