
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances and challenges in designing active site environments in zeolites for Brønsted acid catalysis
Sopuruchukwu
Ezenwa
 and
Rajamani
Gounder
*
and
Rajamani
Gounder
*
Davidson School of Chemical Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: rgounder@purdue.edu
First published on 20th September 2024
Abstract
Zeolites contain proton active sites in diverse void environments that stabilize the reactive intermediates and transition states formed in converting hydrocarbons and oxygenates to chemicals and energy carriers. The catalytic diversity that exists among active sites in voids of varying sizes and shapes, even within a given zeolite topology, has motivated research efforts to position and quantify active sites within distinct voids (synthesis–structure) and to link active site environment to catalytic behavior (structure–reactivity). This Feature Article describes advances and challenges in controlling the position of framework Al centers and associated protons within distinct voids during zeolite synthesis or post-synthetic modification, in identifying and quantifying distinct active site environments using characterization techniques, and in determining the influence of active site environments on catalysis. During zeolite synthesis, organic structure directing agents (SDAs) influence Al substitution at distinct lattice positions via intermolecular interactions (e.g., electrostatics, hydrogen bonding) that depend on the size, structure, and charge distribution of organic SDAs and their mobility when confined within zeolitic voids. Complementary post-synthetic strategies to alter intrapore active site distributions include the selective removal of protons by differently-sized titrants or unreactive organic residues and the selective exchange of framework heteroatoms of different reactivities, but remain limited to certain zeolite frameworks. The ability to identify and quantify active sites within distinct intrapore environments depends on the resolution with which a given characterization technique can distinguish Al T-site positions or proton environments in a given zeolite framework. For proton sites in external unconfined environments, various (post-)synthetic strategies exist to control their amounts, with quantitative methods to distinguish them from internal sites that largely depend on using stoichiometric or catalytic probes that only interact with external sites. Protons in different environments influence reactivity by preferentially stabilizing larger transition states over smaller precursor states and influence selectivity by preferentially stabilizing or destabilizing competing transition states of varying sizes that share a common precursor state. We highlight opportunities to address challenges encountered in the design of active site environments in zeolites by closely integrating precise (post-)synthetic methods, validated characterization techniques, well-defined kinetic probes, and properly calibrated theoretical models. Further advances in understanding the molecular details that underlie synthesis–structure–reactivity relationships for active site environments in zeolite catalysis can accelerate the predictive design of tailored zeolites for desired catalytic transformations.
 Sopuruchukwu Ezenwa | Sopuruchukwu obtained his BS in Chemical Engineering from Tufts University in 2018 and his PhD in Chemical Engineering from Purdue University in 2024 under the mentorship of Prof. Gounder where he investigated the catalytic consequences of active site environments in Brønsted acid aluminosilicates on aromatic alkylation reactions. His research has been recognized by the ChE Faculty Lectureship Award from Purdue (2024), the Richard J. Kokes award from North American Catalysis Society (2022), and the Catalysis and Reaction Engineering Division Travel Award from AIChE (2022). His research interests center on developing molecular-level insights that enable the design of heterogeneous catalysts and chemical processes for producing chemicals and energy carriers from conventional and emerging feedstocks. |
 Rajamani Gounder | Rajamani Gounder is the R. Norris and Eleanor Shreve Professor at the Davidson School of Chemical Engineering at Purdue University. He leads a research group that is recognized for studying the kinetic and mechanistic details of catalytic reactions, for synthesizing and designing zeolites and porous materials with tailored site and surface properties, and for developing methods to characterize and titrate active sites in catalytic surfaces. His group studies the fundamental science of catalysis, and its practical applications to help protect our environment and to develop sustainable, lower-carbon footprint routes to synthesize chemicals and fuels needed for society. |
1. Introduction
Zeolites catalyze a wide range of reactions involved in upgrading hydrocarbons and oxygenates to valuable chemicals and energy carriers.1–4 The substitution of tetrahedrally-coordinated Si4+ in the charge-neutral silica framework ([SiO4/2]) with Al3+ generates anionic centers ([AlO4/2]−), which charge-compensate protons (H+) that serve as active sites.5 The pioneering work of Haag and co-workers demonstrated that the rates (per g zeolite) of a series of catalytic reactions including n-hexane cracking (811 K) were strictly proportional to the Al content (per g zeolite) among MFI zeolites (Si/Al = 15–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000).5,6 Their results suggested either similar reactivity among all 26 possible H+ sites associated with the 12 crystallographic distinct [AlO4/2]− positions in the MFI lattice, or a similar distribution of H+ sites of different reactivity across MFI zeolites synthesized in a similar manner.5,6 Furthermore, isolated Brønsted acid sites in microporous and mesoporous aluminosilicates of diverse topologies have similar acid strength, as rigorously quantified by the deprotonation energy (DPE), or the energy required to remove a proton from the conjugate base ([AlO4/2]−) to non-interacting distances.7–10 However, catalytic diversity exists among varying zeolite topologies and even among samples of a given topology but different synthetic history, as evidenced by variations in site-normalized turnover rates, product selectivity, or deactivation behavior during acid-catalyzed reactions (e.g., alkane cracking11–14 and dehydrogenation,11,15 alkene oligomerization16–19 and hydrogenation,20 alkanol dehydration,21–24 alkanol to hydrocarbons,25–27 aromatic alkylation,28–31 alkanal–alkene coupling32,33).
000).5,6 Their results suggested either similar reactivity among all 26 possible H+ sites associated with the 12 crystallographic distinct [AlO4/2]− positions in the MFI lattice, or a similar distribution of H+ sites of different reactivity across MFI zeolites synthesized in a similar manner.5,6 Furthermore, isolated Brønsted acid sites in microporous and mesoporous aluminosilicates of diverse topologies have similar acid strength, as rigorously quantified by the deprotonation energy (DPE), or the energy required to remove a proton from the conjugate base ([AlO4/2]−) to non-interacting distances.7–10 However, catalytic diversity exists among varying zeolite topologies and even among samples of a given topology but different synthetic history, as evidenced by variations in site-normalized turnover rates, product selectivity, or deactivation behavior during acid-catalyzed reactions (e.g., alkane cracking11–14 and dehydrogenation,11,15 alkene oligomerization16–19 and hydrogenation,20 alkanol dehydration,21–24 alkanol to hydrocarbons,25–27 aromatic alkylation,28–31 alkanal–alkene coupling32,33).
Classical concepts of shape selectivity34–37 were historically used to describe how micropores of varying size and connectivity regulate reactant access to active sites (reactant shape selectivity), control product egress from crystallites (product shape selectivity) and restrict formation of certain transition states (transition-state shape selectivity). Yet, in certain cases where reactivity is not controlled by molecular transport within micropores, these heuristic concepts are limited in explaining catalytic differences among zeolite frameworks possessing similarly sized micropore apertures or in a given zeolite framework containing protons in differently sized voids.38,39 Instead, kinetically controlled reactivity differences during Brønsted acid catalysis are underpinned by the (de)stabilization of reactive intermediates and transition states by electrostatic interactions that depend on the acid strength of the binding site ([AlO4/2]−H+) and by van der Waals interactions that depend on the size and shape of the confining void.38–45 Beyond influences of the binding site and the confining void environment, certain rigid arrangements of proximal anionic Al centers also stabilize cationic intermediates and transition states, conferring additional catalytic diversity to zeolite samples of a fixed framework type, even those such as chabazite (CHA) that contain a single crystallographically unique tetrahedral site (T-site) and similar composition (i.e., Si/Al ratio).13,22,23 Thus, catalytic diversity emanates from variations in the location and arrangement of H+ associated with the siting of framework Al centers at symmetry-distinct T-sites within varying internal (or intracrystalline) micropores of different shapes and sizes (0.4–1.5 nm diameter), at external (or extracrystalline) surfaces and mesoporous environments (>2 nm diameter) (Fig. 1), or in different lattice arrangements.46,47 Furthermore, the void environment and arrangement of Al in zeolites affect the location, speciation, and structure of extra-framework metal cations (Mn+) and complexes (e.g., [MxOy]n+) that are active-site precursors or active sites for metal-catalyzed chemistries (e.g., methane partial oxidation on Cu- and Fe-zeolites48–50 and dehydroaromatization on Mo-zeolites,51–53 alkane dehydrogenation and dehydroaromatization on Ga- and Zn-zeolites,54–56 alkene dimerization and oligomerization on Ni-zeolites,57,58 and reduction and oxidation of nitrogen oxides on Cu-zeolites59,60).
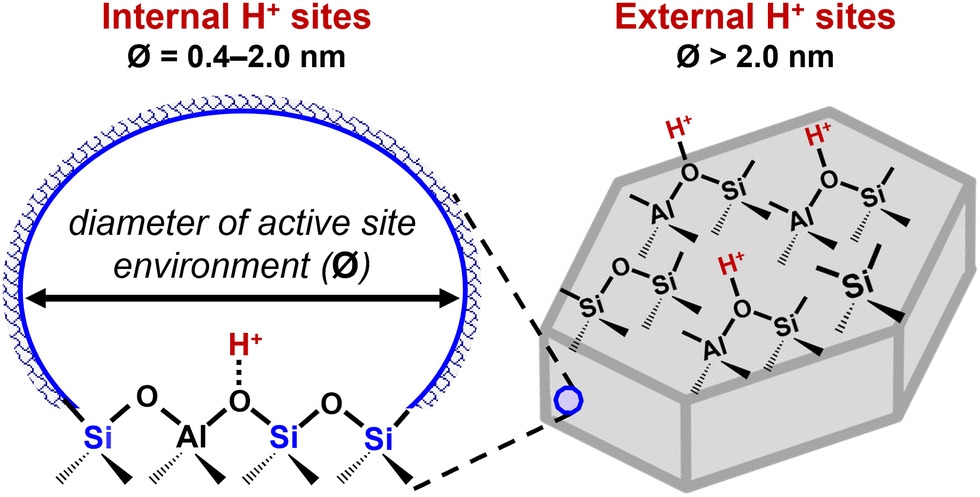 | ||
| Fig. 1 Scheme showing H+ sites in internal micropore environments of varying sizes (0.4–2.0 nm diameter) or at external surfaces and mesoporous environments (>2 nm diameter) of Brønsted acid zeolites. | ||
As highlighted by various reviews and perspectives over the past decade, designing Brønsted acid zeolites for targeted catalytic applications requires strategies to tailor bulk crystallite properties (e.g., crystallite size and morphology, active site density) that influence the intracrystalline diffusion of molecules61–78 and approaches to tailor active site properties (e.g., void environments, local arrangements) that influence the stabilities of reactive intermediates and kinetically relevant transition states.39,46,47,79–87 However, for active site design approaches, challenges in controlling the location of active sites within distinct void environments and in distinguishing their number and intrinsic reactivity limit the ability to tailor zeolites for desired catalytic transformations. This Feature Article focuses on advances and challenges in developing fundamental synthesis–structure–reactivity relationships for Al (and H+) siting within internal confining micropores of varying sizes and shapes and at external unconfined surfaces. We exclude discussions on synthesis–structure–reactivity relationships for Al proximity and arrangement, as our perspective on those topics can be found in a previous publication.47 We also exclude discussions on synthesis–structure–function relationships for bulk crystallite properties that control intracrystalline diffusion phenomena in zeolites.
In Section 2, we connect advances in elucidating the intermolecular interactions that govern the synthetic placement of active sites in distinct internal void environments and highlight complementary approaches to post-synthetically remove acid sites from distinct voids. We also discuss the merits and limitations of characterization techniques often used to assess differences in framework Al siting or H+ void environments. We further describe catalytic insights from selected case studies that delineate how the active site environment alters kinetically controlled reactivity and selectivity for a given zeolite framework with fixed active site density but varying (post-)synthetic origin. In Section 3, we examine (post-)synthetic strategies and characterization techniques to respectively manipulate and quantify the number of active sites at external unconfined environments. We emphasize that, in this focused perspective, the selected examples from previously published works from our group and the broader literature within the past two decades are chosen to illustrate fundamental insights and elucidate molecular-level details underlying synthesis–structure–reactivity relationships for active site environments in zeolite catalysis. In an effort to provide a perspective that is accessible to readers with varying levels of familiarity with the subject matter, we incorporate brief descriptions of some background concepts about zeolites (with references to external resources containing more detailed discussions) that are helpful to understand the insights presented in this Feature Article. We conclude in Section 4 by providing our outlook on knowledge gaps and future directions that can advance the design of zeolites with tailored active sites for targeted catalytic applications. Throughout this perspective, we underscore our viewpoint that the close integration of precise (post-)synthetic control strategies, validated characterization techniques, well-defined kinetic probes, and properly calibrated theoretical models is important in developing fundamental synthesis–structure–reactivity relationships that underpin the catalytic diversity of active site environments in Brønsted acid zeolites.
2. Active sites within distinct internal void environments of a fixed zeolite framework
The siting of Al at crystallographically distinct lattice positions within zeolite micropores dictates the location of their attendant protons within confining voids of varying sizes and shapes and, in turn, influences the stability of kinetically relevant intermediates and transition states. The structural diversity of zeolites requires brief descriptions of some common metrics used to distinguish relevant void features. The connection of TO4/2 building units (T = Si or Al) forms rings (denoted by X-MR, where X is the number of T-atoms members) and void features such as cavities and channels, which are used to classify zeolite frameworks by the number of T-atoms delimiting their pore apertures into small (8-MR), medium (10-MR), and large (12-MR) pore zeolites.88,89 The pore limiting diameter (PLD) and the largest cavity diameter (LCD) describe the diameter of the largest sphere that can diffuse through the microporous structure and that can be circumscribed inside zeolitic cavities, respectively.90–92 In many cases, void shapes can be described by the maximum and minimum diameters of the cross-section (e.g., 0.51 nm × 0.55 nm for MFI sinusoidal channels),89 while pore dimensionality can be used to describe the ability of guest molecules to diffuse along up to three independent directions (1D; 2D; 3D).88,89 Further discussions of relevant void and topological features of zeolites can be found elsewhere.88–98 In this Feature Article, most of our discussions focus on the FER, MFI and MOR zeolite frameworks (Fig. 2). | ||
| Fig. 2 Void and topological features of three key zeolites (FER, MFI, MOR) discussed in this work. The pore dimensionality and some ring features (8-MR, 10-MR, 12-MR) are indicated. Structures obtained from IZA database.89 | ||
In this section, we discuss the intermolecular interactions between the organic structure directing agent (SDA) and zeolite framework that underlie synthetic protocols to selectively place Al in desired void environments and the complementary post-synthetic strategies to selectively remove H+ from undesired void environments. We further describe experimental characterization techniques to probe active site locations within distinct zeolite voids. We incorporate discussions of computational assessments that are valuable in developing molecular-level insights into the (post-)synthetic strategies and characterization techniques. We then conclude by using two probe reactions to demonstrate how the catalytic consequences of distinct Al (and H+) environments of a fixed zeolite framework are elucidated using mechanistic interpretations of rates measured under conditions of strict kinetic control and interpreted alongside theoretical calculations.
2.1. Strategies to control active site placement within distinct void sizes of a fixed framework
 | ||
| Fig. 3 Roles of organic SDA during zeolite synthesis: (A) stabilizing the crystallization from amorphous precursors via van der Waals interactions with silicate species. Adapted from Burkett et al.101,102 (B) Stabilizing the siting of Al at lattice positions via electrostatic interactions only (for quaternary organic SDA) and, in some cases, via additional hydrogen bonding interactions (in the case of non-quaternary organic SDA). | ||
Early experimental and theoretical studies recognized how Coulombic interactions influence Al siting at T-sites closest to the charged N+ center of the organic SDA during synthesis. Using advanced NMR techniques, Shantz et al.113,114 established that during the synthesis of MTW (a 1D large pore zeolite), the cationic N+ center of the benzyltrimethylammonium organic SDA is preferentially ordered near the anionic framework Al center of the zeolite lattice, consistent with Coulombic type interactions. From these results, they suggested that knowledge of the geometric arrangement of organic SDAs in the zeolite micropores and proper selection of organic SDAs with different charge distributions should be a useful starting point to tune the siting of trivalent framework heteroatoms (e.g., Al) during zeolite synthesis.113,114 Sastre et al.115,116 used molecular mechanics simulations on ISV (a 3D large pore zeolite with five distinct T-sites) and MEI (a 3D large pore zeolite with four distinct T-sites) to show that Coulombic interactions are maximized (or electrostatic energy minimized) when anionic Al are sited at lattice positions closest to the N+ center of the organic SDA and that these Coulombic interactions have a greater influence on Al siting than the intrinsic stabilities of Al substitution at distinct T-sites in absence of organic SDA.
Later studies by Pinar et al.117 and Román-Leshkov et al.118 have provided a well-known example of how organic SDAs can bias the relative proportion of H+ within the smaller 8-MR voids (H8-MR+; 0.48 nm × 0.35 nm)89 and the larger 10-MR voids (H+10-MR; 0.54 nm × 0.42 nm)89 of FER, a 2D medium-pore zeolite with four distinct T-sites (Fig. 4A). As summarized in Fig. 4B, the synthesis of FER (Si/Al = 10–17) with varying organic SDAs (and combinations thereof), resulted in significant differences in the fraction of H+ in the 10-MR (H+10-MR/H+total = 0.11–0.73).117,118 From Rietveld refinement of powder X-ray diffraction (XRD) data accompanied by molecular mechanics simulations, tetramethylammonium (TMA+) was found to reside exclusively in the FER cage (accessibly only by 8-MR channel) (Fig. 4), whereas pyrrolidine (Pyrr) or its protonated form (pyrrolidinium; Pyrr+) can reside within either the 10-MR channels or FER cage.119,120 Molecular mechanics simulations reveal that, in addition to electrostatic interactions with the [AlO4/2]−, Pyrr+ can engage in hydrogen bonding interactions with the anionic oxygen of the [AlO4/2]− (Fig. 4), which results in a stronger influence of Pyrr+ over TMA+ on the Al siting in the lattice, especially when Pyrr is occupying the FER cavity.121 These result in synthesis with Pyrr as the sole organic SDA having the lowest fraction of H+10-MR (H+10-MR/H+total = 0.11), while the addition of TMA+ to the synthesis media displaces some Pyrr from the FER cavity to the 10-MR channel and results in slightly higher fractions of H+10-MR (H+10-MR/H+total = 0.16).117,119 Bulkier organic co-SDAs (>0.50 nm diameter) such as 1-benzyl-1-methylpyrrolidinium (BMP+)122,123 or hexamethyleneimine (HMI)118 are restricted to 10-MR channels because of size exclusion from the smaller FER cage, and this results in increased fractions of H+10-MR. Furthermore, synthesis using HMI results in the highest fraction of H+10-MR (H+10-MR/H+total = 0.73) because hydrogen bonding interactions of the NH2+ group and less shielding by the R2 group (R = alkyl) permit stronger interaction with [AlO4/2]− in 10-MR relative to the bulkier quaternary alkylammonium structure of BMP+.117,118 More recent studies by other groups124,125 have provided further examples of organic SDAs (morpholine, piperidine, ethylenediamine, pyridine, cyclohexylamine, and 1,6-diaminohexane) that lead to different biases in Al siting in synthesized FER zeolites. Thus, for FER zeolite containing four distinct T-sites with bridging framework O (Of) atoms that can be oriented in either 8-MR or 10-MR voids, the size and charge distribution (e.g., R4N+vs. R2NH2+) of the organic SDA dictates the magnitude of its intermolecular interaction with framework anionic Al centers, which determines the siting of Al (and H+) within distinct void environments (Fig. 4).
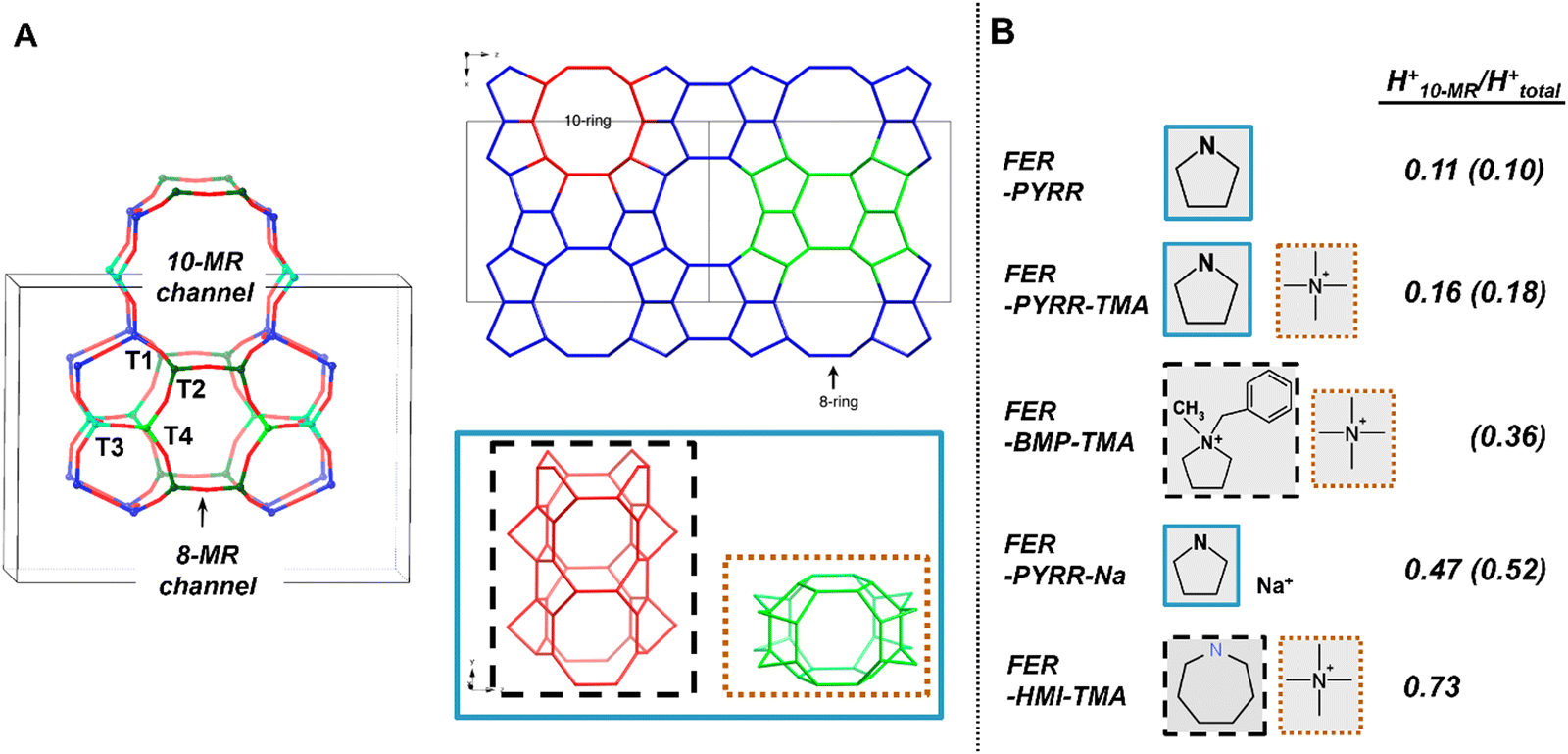 | ||
| Fig. 4 (A) FER pore structure showing the four distinct T sites, the 10-MR channels, and the ferrierite cavity accessible from 8-MR pore openings. T4 is accessible only from the 8-MR channel. Adapted with permission from Pinar et al.126 Copyright 2021 American Chemical Society and from Pinar et al.119 Copyright 2013 American Chemical Society. (B) Organic SDA used to synthesize FER zeolites and their influence on the H+ void environment in synthesized FER zeolites. Data for H+10-MR/H+total obtained from Pinar et al.117,119 (shown in parentheses) and Román-Leshkov et al.118 The organic SDAs are color coded by the FER voids that they reside in (A). | ||
In contrast to FER, which contains four crystallographic distinct T-sites in a 2D topology of intersecting and co-planar 8-MR and 10-MR channels, MFI is a lower symmetry framework containing 12 crystallographically distinct T-sites (orthorhombic phase) in a 3D topology of intersecting 10-MR straight (0.53 nm × 0.56 nm) and 10-MR sinusoidal (0.51 nm × 0.55 nm) channels (Fig. 5A) which lie in orthogonal planes that allow for molecular diffusion along the three crystallographic directions.89,127,128 More importantly, the 12 distinct T-sites of MFI result in 26 distinct neutral Of atoms for the silicate form (i.e., ≡Ta–O–Tb ≡, where Ta = Tb = Si) or 48 distinct anionic Of atoms for aluminosilicate form (i.e., ≡Ta–O−–Tb ≡, where Ta ≠ Tb = Si or Al) and these bridging anionic Of can have attendant protons that reside within the channels (∼0.55 nm diameter)92 or their larger intersections (∼0.70 nm diameter).92,129,130 The increased structural complexity of MFI zeolite framework (relative to FER) present various synthetic challenges to precisely tune the void location of the proton. To illustrate, each isolated Al-substituted T-site of MFI has four possible bridging Of–H that are isoenergetic (i.e., DPE within 10 kJ mol−1)7 and can host protons in either channel or intersection void environments, with the exception of T4 site that can only host protons accessible in the sinusoidal channel environments.130
 | ||
| Fig. 5 (A) MFI pore structure showing the sinusoidal channels (blue; across a–c plane), straight channels (blue; along b-axis), and the channel intersections (green) (B) Conventional organic SDA (TPA+) and nonconventional organic SDA mixtures (EDA + TPA, DABCO + MA) used to synthesize MFI zeolites. (C) Energies of Al siting in positions T1–T11 (relative to T12) with varying organic SDA complexes: D-W = [DABCO-H-H2O]+, D-M = [DABCO-H-MA]+; (S) indicates calculation performed with an implicit solvation model. Reproduced with permission from Ezenwa et al.131 Copyright 2024 American Chemical Society. | ||
The synthesis of MFI zeolites using the conventional tetra-n-propylammonium (TPA+)132 SDA (Fig. 5B) is known to preferentially position [AlO4/2]− in T-sites bordering the larger intersections and that are closest to the N+ center of the TPA+ molecules that are fixed at MFI intersections, because electrostatic interactions are maximized when the N+–Al− distance is minimized.130,133,134 Yokoi and colleagues135–138 reported that the synthesis of MFI zeolites (Si/Al = 20–50) using mixtures of alkylamine (e.g., cyclohexylamine) or alcohol (e.g., pentaerythritol, PET; trimethylolethane, TME) molecules and Na+ resulted in increased siting of Al at T-sites within the more constrained channel environments because neutral branched organic molecules occupy intersection voids, thereby forcing Na+ to charge compensate Al centers (Na+/Al = 0.3–1.1)135,137 at channel environments. We note that prior to these reports,135–137 MFI zeolites had been synthesized with >30 different types of organic molecules132,139–157 but with scant reports79 of the resulting consequences on Al siting within different voids during synthesis with non-conventional organic SDA. Further studies by other research groups158–162 also reported or inferred that MFI synthesized using the earlier reported alcohol molecules (PET, TME)136,137 or n-butylamine (NBA)158,160 resulted in increased siting of Al at the channel environments relative to the intersection environments that are favored by TPA+. A recent study by Yokoi and colleagues163 further proposed that PET molecules prevented the interaction of TPA+ molecules and [AlO4/2]− centers thereby enhancing the ability of Na+ to place Al centers in channels during synthesis of MFI using mixtures of TPA+, PET and Na+. However, detailed molecular-level insights into the influence of organic SDA structure on Al siting remain limited for MFI zeolites.
Recently, we reported131 that the synthesis of MFI zeolites of fixed Al content (Si/Al ≈ 50) using an either an equimolar mixture of 1,4-diazabicyclo[2.2.2]octane (DABCO) and methylamine (MA)164 or a mixture of excess ethylenediamine (EDA) with minor quantities of TPA+,165 resulted in significant bias of Al towards the more confined channels. In contrast, the synthesis of MFI zeolites using TPA+ as the only SDA resulted in the predominant siting of Al within the larger intersections, in agreement with density functional theory (DFT) studies.130,133 In the case of MFI crystallization using DABCO/MA mixtures, DFT simulations reveal that the ability of protonated DABCO complexes to reorient within MFI intersections and participate in additional hydrogen-bonding interactions with [AlO4/2]− stabilizes a broader range of Al substitution locations and facilitates the placement of Al at T sites in the smaller channel environments that are less favored by TPA+ (Fig. 5C).131 We surmised that a similar flexibility and ability of protonated EDA to form H-bonding interactions biases the Al positioning away from the intersection-dominant locations preferred solely by TPA+.131 Indeed, earlier studies by Perego and colleagues148,149 reported that EDA molecules are stabilized by favorable van der Waals interactions in the smaller channels of MFI and that the compositional N/B ratio ∼1 in samples crystallized from borosilicate solutions implies that EDA interacts with framework B either via electrostatic interactions in their protonated form or via N⋯H–Of hydrogen bonds in their unprotonated form,148,149 suggesting a similar mechanism by which EDA may bias the siting of trivalent heteroatoms towards channels. Furthermore, the findings from our group130,131 are consistent with those of a recent DFT and molecular dynamics (MD) study that proposed that organic SDAs with different charge distributions and mobility within voids can alter the Al siting towards T-sites that are further away from MFI intersections.133 Together, these molecular-level details depict a mechanistic link between organic SDA structure and Al siting in MFI zeolites.
Seldom examined in prevailing discourses on organic SDA influence during zeolite synthesis, H-bonding interactions have been proposed to play a significant role in the assembly of organic guest molecules within diverse inorganic microporous hosts (e.g., aluminosilicates, aluminophosphates, zincophosphates, gallogermanates)121,166–171 For instance, H-bonding interactions between tris(ethylenediamine)cobalt(III) complexes ([Co(en)3]3+) and aluminophosphate frameworks were reported to promote the crystallization of aluminophosphates and transfer the chirality of the metal complex to the inorganic hosts.166,167,170 Furthermore, Gómez-Hortigüela and colleagues used molecular simulations to identify that strong H-bonding interactions between flexible organic molecules containing H-bond forming groups (–NH or –OH) and the Of atoms bridging isomorphically substituted lower-valent dopants (e.g., Mg2+ for Al3+ in AFI aluminophosphates168,169 or Al3+ for Si4+ in FER aluminosilicates119,121) drive the siting of dopants in the framework.172 These insights demonstrate that a computationally informed choice of organic molecule capable of participating in hydrogen bonding interactions with the zeolite framework may be an important design strategy to place active sites in desired environments.131
Further notable examples of the influence of organic SDAs on framework Al siting for a fixed zeolite framework can be found in reports for (a) MOR, a 1D large-pore zeolite containing four distinct T-sites distributed among 8-MR side-pockets (0.57 nm × 0.26 nm; 0.48 nm × 0.34 nm) and 12-MR channels (0.70 nm × 0.65 nm),173–177 (b) RTH, a 2D small-pore zeolite with four distinct T-sites distributed among non-distorted 8-MR (0.41 nm × 0.38 nm) and distorted 8-MR (0.56 nm × 0.25 nm) pore apertures,178 (c) MWW, a 2D medium-pore zeolite containing eight distinct T-sites (up to 14 T-sites for SSZ-70) distributed among 10-MR sinusoidal channels within layers (0.51 nm × 0.41 nm) and 12-MR supercages (0.71 nm × 0.71 nm) with 10-MR apertures between layers (0.55 nm × 0.40 nm),179,180 and (d) IFR, a 1D large-pore zeolite containing four distinct T-sites within the 12-MR channel (0.62 nm × 0.72 nm).181
Together, experimental and computational studies demonstrate that beyond intrinsic (i.e., thermodynamically preferred under SDA-free conditions) Al siting preferences at crystallographic distinct T-sites and van der Waals interactions that stabilize organic SDAs within specific pore geometries, intermolecular interactions (e.g., electrostatics, hydrogen bonding) between organic SDAs and Al centers play a major role in the placement of framework Al at distinct T-sites; such interactions depend on the size, structure, chemical functionality, and charge distribution of organic SDAs and their mobility within the voids during crystallization.
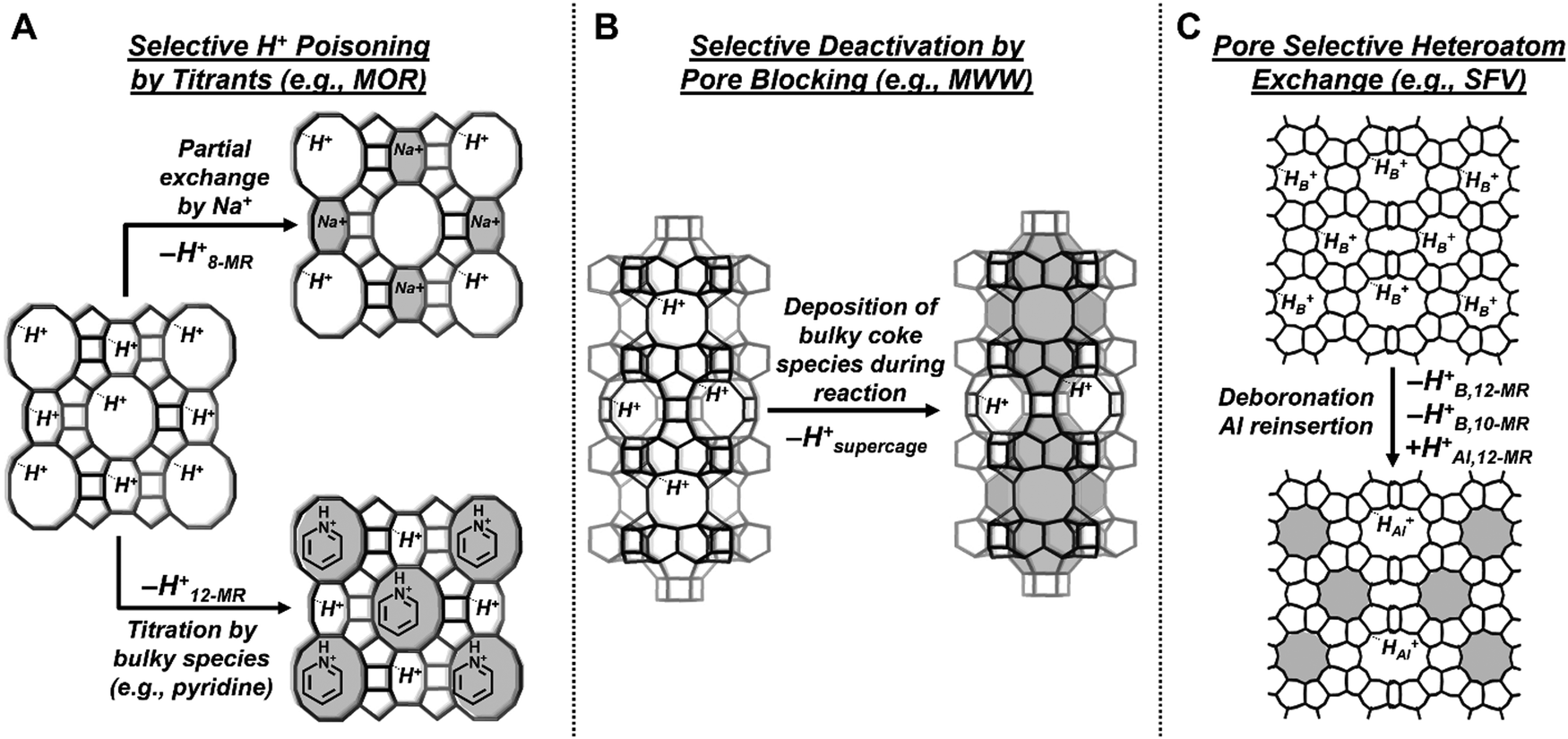 | ||
| Fig. 6 Schematic summarizing the highlighted post-synthetic techniques to modify Al and H+ siting within distinct micropore environments of zeolites. (A) Selective poisoning of H+ sites in 8-MR and 12-MR of MOR by Na+ or bulky titrants, respectively. (B) Selective deactivation by pore blocking of H+ sites in 12-MR supercages of MWW zeolites. (C) Selective heteroatom exchange in SFV zeolites. Shaded regions depict voids containing inactive sites. | ||
The post-synthetic treatment of MOR zeolites in aqueous Na+ solutions at sub-unity Na+ to Al molar ratios is a well-known technique to preferentially exchange H+ in the 8-MR side pockets (H8-MR+) with catalytically inactive Na+ while preserving H+ in 12-MR channels (H12-MR+),11,182–185 because of closer contacts of Na+ cations with framework oxygen atoms within smaller 8-MR voids than those within larger 12-MR voids.11 On the other hand, H12-MR+ are preferentially poisoned by pyridine or alkylpyridines (e.g., 2,6-dimethylpyridine),174,185–187 exchanged by tetramethylammonium ions,188 or eliminated via silylation with trimethylchlorosilane189 because size constraints exclude these bulky species from accessing H8-MR+. Furthermore, as surmised by a recent experimental and theoretical study13 from our group that found that Na+ preferentially titrated H+ in 6-MR Al–Al pair configurations over H+ at isolated Al sites in CHA zeolites, strong effects of preferential Na+ titration of H+ within distinct void environments of aluminosilicates may reflect an underlying influence of Al–Al pairs. Thus, the selective titration of H+ sites is a post-synthetic approach to tune the H+ void environments in certain zeolites.
The selective deposition of unreactive organic residues in specific voids provides another established strategy to post-synthetically tune H+ siting in zeolites with independent non-connected pore systems. During the cracking of n-hexane and 3-methylpentane (588 K) in MOR, H12-MR+ were preferentially deactivated with time-on-stream when compared to H8-MR+.190 Similarly, H+ located within 12-MR supercages of MWW zeolites (0.71 nm × 0.71 nm) were preferentially deactivated with time-on-stream over H+ located within the 10-MR sinusoidal channels (0.51 nm × 0.41 nm) during n-hexane and 3-methylpentane cracking (588 K),190,191n-heptane cracking (623–723 K),192–194m-xylene transformation (623 K),195,196 toluene methylation (623 K),30,31 methanol to hydrocarbons (723 K),197,198 and syngas to gasoline (633 K).199 However, for certain zeolite topologies with interconnected pore systems (e.g., MFI), deposition of bulky organic residues imprecisely alters the Al siting and may introduce unintended collateral effects, given that blocking of H+ sites in intersections will restrict access to H+ sites in the connected channels and vice versa. Nevertheless, the selective deposition of coke species remains a viable technique to post-synthetically eliminate H+ sites within distinct void environments of certain zeolites containing non-intersecting pore systems.
Selective exchange of trivalent heteroatoms of different reactivities in distinct voids of zeolites is an emerging design strategy to post-synthetically introduce Al to specific void environments of zeolites. For example, H+ that charge-compensate framework boron (HB+) are typically catalytically irrelevant relative to HAl+ because the DPE of HB+ is ∼70 kJ mol−1 higher than that of HAl+.200–203 The synthesis of borosilicate zeotypes followed by deboronation and subsequent Al reinsertion (B → Al exchange) has historically provided an indirect route to obtain certain aluminosilicate zeolites that are inaccessible from direct crystallization using amorphous Al reagents.204–206 Beyond synthesizing new zeolite frameworks, Zones, Koller and colleagues further observed that (i) in certain zeolites, B atoms typically site at structural motifs such as 4-ring chains,207,208 (ii) hydrated Al3+ cations are unable to access 10-MR pores,207,209 (iii) heteroatom siting is preserved during B/Al exchange without significant T-site or silanol nest migration.207,208 These observations suggest that B → Al exchange might be a viable strategy to tune Al siting in certain zeolite frameworks, a process referred to as “pore selective aluminum reinsertion”.206 This was demonstrated for post-synthetically B → Al exchanged SFV (SSZ-57) zeolites that contained Al in only 12-MR pores209 as opposed to directly synthesized Al-SFV zeolites that contained Al in both 12-MR and 10-MR pores.210,211 Such emerging techniques to selectively reinsert Al into specific lattice positions hold promise, but find limited applications for medium-pore zeolites as reported earlier207,209 and observed in our own unpublished studies. Alternative strategies such as gas-phase Al exchange in moisture-free atmospheres or liquid-phase exchange in moisture-free solvents212 into deboronated zeolites could be explored to overcome the current hurdles for medium-pore zeolites and allow the indirect synthesis of aluminosilicates with tailored Al siting than would have been obtained if synthesized directly in Al-containing form.
2.2. Experimental characterization of Al siting within distinct voids
Synthetic and post-synthetic strategies to tune the siting of Al (and attendant H+) within distinct voids of zeolites require characterization techniques to assess the resulting impact on Al siting within distinct voids. These techniques are broadly distinguished by those that seek to identify the lattice position of framework Al and those that seek to determine the void environment of the protons directly without adsorbed probes or indirectly using adsorbed probes.Solid-state 27Al magic angle spinning nuclear magnetic resonance (MAS NMR) spectra are sensitive to differences the local coordination environments of 27Al nuclei and are widely used to distinguish tetrahedrally coordinated framework Al (∼54–65 ppm) from octahedrally coordinated extra-framework Al species (∼0 ppm).213 The early reported correlation by Lippmaa et al.214 of 27Al chemical shifts with the average Al–O–Si bond angles in various Al-rich aluminosilicates (Si/Al = 1–5) suggests that 27Al MAS NMR may be able to capture subtle differences in the local structure of framework Al due to differences in Al siting among distinct lattice positions. Han et al.215 reported that two observed peaks (∼51.2 and ∼53.5 ppm) in both the 27Al MAS NMR and the two-dimensional 27Al triple-quantum (3Q) MAS NMR spectra of MFI zeolites (Si/Al = 14–250) result from overlapped signals of two subsets of the 12 distinct T-sites, and further used the correlation by Lippmaa et al.214 and Al–O–Si angles from the crystallographic data of Olson et al.128 to estimate isotropic 27Al chemical shifts for the 12 distinct T-sites of orthorhombic MFI.
Subsequent studies by Sklenak et al.216–218 combined 27Al 3Q MAS NMR experiments and quantum mechanics molecular mechanics (QM/MM) simulations to perform a partial assignment of observed resonances to specific T-sites in monoclinic MFI (24 distinct T-sites). They noted that no simple correlation exists between 27Al isotropic chemical shifts and average calculated Al–O–Si angles and that the crystallographic X-ray data contained artefacts in Al–O–Si bond angles because of low Al content. Nevertheless, studies by Han et al.,215 Sklenak et al.,216–218 and by other groups (including ours)130,131,134,137,219,220 agree on the non-random siting of Al at distinct T-sites of MFI zeolites synthesized using TPA+. However, across these studies on MFI and on other low symmetry zeolites containing >4 distinct T-sites,179,221–225 the majority (>90%) of the isotropic 27Al chemical shifts fall within a narrow range (<10 ppm)219 and result in experimental 27Al signals that are still too close to be unambiguously resolved despite advances in multiquantum (MQ) MAS experiments and increases in NMR resolution in recent years.226,227 On the other hand, for zeolite topologies (FER,125,228,229 RTH,178 IFR181) of higher symmetry containing 4 distinct T-sites, the 27Al 1D or 2D MQ MAS NMR spectra can be partially resolved into at least two components that significantly change with varying synthesis organic SDA at fixed solid Si/Al ratios (Fig. 7).
 | ||
| Fig. 7 Solid-state 1D 27Al MAS NMR spectra of (a) MFI, (b) FER, (c) RTH zeolites synthesized using varying organic SDA. Adapted with permission from Ezenwa et al.,131 Pinar et al.,228 and Liu et al.,178 respectively. Copyright 2024 American Chemical Society, 2014 Elsevier, 2014 Royal Society of Chemistry The spectra show qualitative differences which suggest that the various OSDA biased locations of framework Al among various lattice positions. | ||
Besides NMR techniques, X-ray diffraction (XRD) methods have been applied to deduce the lattice positions of framework heteroatoms (e.g., Al, B) through locating the positions of well-ordered organic SDA molecules in as synthesized zeolites.230 In this approach, the single-crystal or powder XRD data are processed using Rietveld refinement231 to obtain positional parameters for the atoms of the inorganic zeolite host and the organic SDA guest. The refined interatomic distances between the C and N of the organic SDA and the Of atom are used alongside interaction energies calculated using molecular simulations (MM or MD) to draw conclusions on the highest probability locations of the organic SDA and the anionic Al centers bearing Of that are closest to the N+ center of the organic SDA. This approach has been partly successful in determining the preferred lattice positions of framework Al in FER zeolite synthesized using varying organic SDA (described in Section 2.1.1)119,120,124 and of framework B in various zeolite frameworks.230,232,233 In addition to challenges in distinguishing framework Si from Al because of their similar scattering factors,119,120 structural refinements of powder XRD patterns are also limited by lower contrast of organic guests because of their lighter scattering relative to the inorganic host, low symmetry of occluded organic SDA, and low resolution of intensities from overlapping diffraction planes.230
Techniques such as simulated annealing,234 a systematic global optimization routine, have been used to improve the extraction of locations of organic SDA and framework heteroatoms from structural refinement of powder XRD patterns.230,232 In addition, XRD data collected at the Al K-edge, where the Al scattering factor significantly diverges from that of Si because of resonant scattering (termed anomalous X-ray diffraction), has been combined with conventional powder XRD data to unambiguously and quantitatively determine the occupancies of Al at the four distinct T-sites of two H-form FER zeolites synthesized using different organic SDAs (Fig. 8).126 Thus, powder XRD data when obtained at the Al K-edge126 or when refined using simulated annealing230 could enable locating the position of framework Al in lower symmetry zeolite frameworks (e.g., MFI). Additional X-ray based techniques such as extended X-ray absorption fine structure (XAS) spectroscopy,223,224 Al valence-to-core X-ray emission spectroscopy (XES)235 and fluorescence spectroscopy using X-ray standing waves (XSW)236 have also been used to estimate the occupation of distinct T-sites by Al in BEA (9 T-sites), FER (4 T-sites) and NAT (2 T-sites) zeolite, respectively.
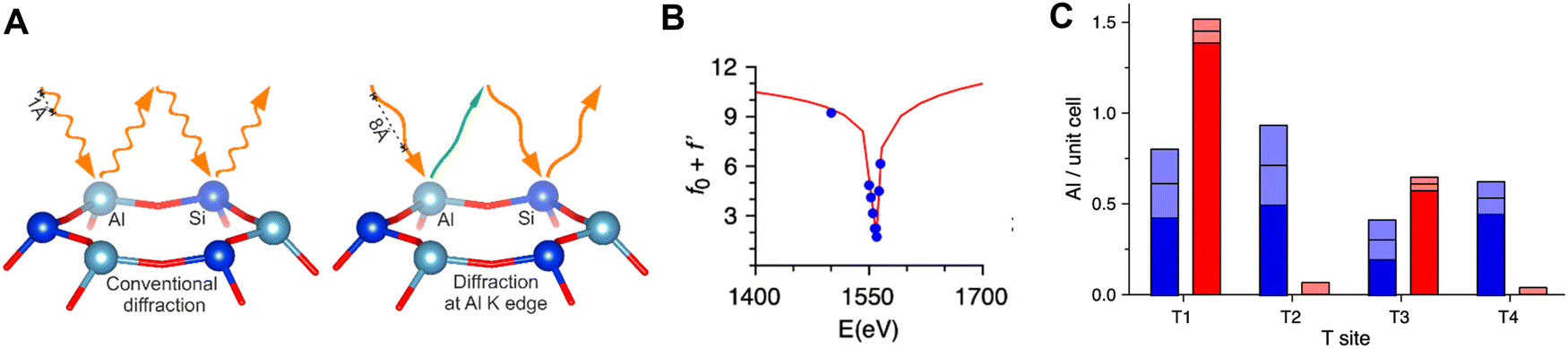 | ||
| Fig. 8 (A) Illustration of anomalous X-ray scattering at the Al K edge (1.56 keV or 7.95 Å) as an approach to distinguish the similar scattering factors of Al and Si within FER zeolites containing Si/Al ratios >5. (B) The real part (f0 + f′) of the X-ray scattering factor (f = f0 + f′ + if′′) as a function of the X-ray energy. Within the vicinity of the Al K edge, the f′ and f′′ become more significant than f0 (the number of electrons) which changes the scattering power of Al. (C) Distribution of Al among four distinct T-sites in two FER zeolites (2.2 Al per unit cell) of varying synthetic origins. Reproduced with permission from Pinar et al.126 Copyright 2021 American Chemical Society. | ||
In contrast to techniques used to identify the lattice positions of framework Al, alternative approaches measure differences in H+ environments, which are then mapped to the most probable framework Al positions, given knowledge of void environments of their bridging Of atoms and assumptions of isoenergetic distributions of associated H+. During infrared (IR) spectroscopy of H-form zeolites, the Of–H+ bond stretching frequencies (νOH) for Brønsted acid sites are known to be sensitive to void environments as reflected in a wide range of νOH (3550–3660 cm−1) across zeolites of varied framework topologies that contain differently sized void environments.7,237–246 In addition, reported molar extinction coefficients for Brønsted acid sites (εOH) are either similar or vary (2–20 cm μmol−1) with void size.183,238,245–248 These variations have been broadly ascribed to Of–H+ bonds perturbed via interactions with nearby Of atoms regardless of the specific nature (electrostatic,241 hydrogen bonding,242–244,248–250 or van der Waals7), where smaller voids offer more intimate interactions that lengthen Of–H+ bonds and decrease νOH.7,241–244
The IR spectra of various H-MOR zeolites with a narrow composition (Si/Al = 6–10), but varied commercial origins, were found to consist of an asymmetric IR band in the Brønsted acid Of–H+ stretching region which can be deconvoluted into two symmetric bands arising from H8-MR+ (∼3590 cm−1) and H12-MR+ (∼3610 cm−1) (Fig. 9A).11,182–186 Among these MOR samples, the ratios of H8-MR+ and H12-MR+ to H+total varied with sample origin11,182–186 and with selective poisoning of either H8-MR+ by Na+ cations11,182–185 (Fig. 9B) or H12-MR+ by (alkyl)pyridines;185,186 these indicate that the unreported synthetic origins resulted in non-random siting of Al among the four symmetry distinct T-sites and, in turn, non-random distribution of H+ among the 10 distinct Of atoms. Recent studies on MOR zeolites have further reported that varying organic SDA at fixed Al and total H+ contents biases the relative proportions of H8-MR+ and H12-MR+, measured via deconvolution of Of–H+ IR spectra.175,176 However, across various research groups, quantitative assessments of H+ locations within distinct MOR voids vary with spectral deconvolution procedures that differ in numbers of primary components and in optimization routines for spectral features (e.g., peak shape and positions).175,176,183,185,251 In addition, values of εOH and νOH depend not only on intermolecular interactions with the nearby void environment7,241–244 but also on the local configurations of proximal H+ that are sensitive to temperature even for high-symmetry zeolites containing one crystallographic distinct T-site (e.g., FAU,252 CHA13,253).
 | ||
| Fig. 9 (A) MOR framework structure showing the four distinct T sites and some framework O sites. T3–O31 and T3–O33 positions are located exclusively in the 8-MR side-pockets. The rest of the T–Of pairs are either located within the 12-MR channel or at the boundary between the 8-MR side pockets and the 12-MR channels. Adapted with permission from Boronat et al.254 Copyright 2008 American Chemical Society (B) Deconvolution of OH IR spectra of on a series of partial Na+-exchanged MOR zeolites (Si/Al = 10; (a)–(f) reflects increasing degree of Na exchange (Na/Al = 0.00, 0.17, 0.27, 0.41, 0.55, 0.90)). Reproduced with permission from Bhan et al.185 Copyright 2007 American Chemical Society. | ||
The accessibility of probe molecules to distinct voids of certain zeolites has been used to quantify differences in H+ environment.244,255 Specifically, the adsorption of pyridine (kinetic diameter, dkin ∼ 0.58 nm) on H-form zeolites followed by quantification using IR or NMR spectroscopy is a common procedure for quantifying H+ sites in pores with apertures larger than those of 8-MR (∼0.40 nm diameter).238,244,255 This approach has enabled the quantification of H12-MR+ of MOR zeolites182,185,186 and H+10-MR of FER zeolites117,118 because pyridine cannot access H8-MR+. However, reproducible quantification for certain zeolites can be limited by partial accessibility of pyridine to H+ sites because of slow diffusion rates during the timescale of the experiments and by variations in molar extinction coefficients for the ring vibration bands or the N–H+ stretching bands of pyridinium ions.255–257 In addition, for zeolites containing distinct but interconnected pore environments (e.g., ∼0.55 nm channels and ∼0.70 intersections of MFI), pyridine indiscriminately titrates all H+ sites, thus preventing the quantification of distinct site environments.
The constraint index (CI) test,258 given by n-hexane-to-3-methylpentane cracking rate ratio (typically between 623–673 K), is one of many catalytic probe reactions259 that have been historically developed to assess differences in void features (limiting aperture size and void connectivity) of zeolite frameworks. In cases where reactivity is weakly influenced by intracrystalline diffusion limitations, the CI test may also sense the influence of active site environments on the stabilities of differently sized transition states.258,259 However, CI test values deviate from expected trends for certain zeolite topologies,190,191 are sensitive to presence of independent pore systems that deactivate at different rates (e.g., MOR and MWW),190 and vary significantly with temperature258 for medium-pore zeolites because of competition either between enthalpic and entropic effects260 or between monomolecular and bimolecular mechanisms.261,262 Nevertheless, MFI synthesized using certain non-conventional organic molecules (e.g., PET, TME, NBA) were reported to have higher CI test values relative to MFI samples synthesized using TPA+, which was used to infer that non-conventional MFI samples contained significant numbers of active sites within the more constrained channel environments.135,137,158–160,163
Together, the merits and drawbacks of the highlighted characterization techniques for assessing differences in framework Al siting or in H+ void environments pinpoint areas to advance the development of improved techniques that unambiguously quantify active sites within distinct void environments, even among lower symmetry frameworks such as MFI and MWW zeolites.
2.3. Catalytic consequences of acid site location in distinct void environment of a given zeolite
Under conditions where catalysis is not influenced by constraints on molecular diffusion within micropores, reactivity and selectivity differences across zeolites reflect the effects of pore confinement on the stabilities of intermediates and transition states at active sites within voids environments of varying sizes and shapes.38,39 Confinement effects reflect attractive van der Waals interactions between bound species and confining voids that offset the energy penalties associated with the structural distortions required to accommodate organic guests within inorganic host and the entropic losses of bound species upon confinement.32,43,44,263–265 Furthermore, confinement effects become more significant when transition states and their precursors or other competing transition states are solvated to different extents because of differences in sizes.39 Elucidating the catalytic consequences of active site environments requires maintaining the link between reactivity and the local structures of active sites and bound species;38 this necessitates measurement of rates and selectivity in absence (or accounting for) intracrystalline diffusion constraints while minimizing the contributions of side reactions that often convolute kinetic, thermodynamic and transport phenomena.44,77,266–268 There are many examples of complex reaction networks that are reported to be sensitive to differences in active site environments in a given zeolite (e.g., methanol to hydrocarbons in MFI, MEL, MWW137,159,197,269–271), but for the aforementioned reasons, we do not discuss those in this perspective. Rather, in this section, we use the methylations of carbon monoxide (CO) and toluene by DME at low reaction temperatures (<473 K) to demonstrate how the active site environment alters kinetically controlled reactivity and selectivity for a given zeolite framework with fixed active site density but of varying synthetic origin. | (1) |
 | ||
| Fig. 10 (A) Methyl acetate formation rates (per g Al) as a function of CO pressure during CO methylation (438 K) by DME (2–16 kPa) on H-MOR zeolite (Si/Al = 10). Solid lines represent regressed best fit to eqn (1). Adapted with permission from Cheung et al.275 Copyright 2006 Wiley-VCH (B) Methyl acetate synthesis rate (per g zeolite; 438 K, 930 kPa CO, 20 kPa DME) as a function of number of H+ in 8-MR of Na,H-MOR and H-FER zeolites (per g zeolite). Dashed line represent line of best fit for MOR data. Adapted with permission from Bhan et al.185 Copyright 2007 American Chemical Society (C) Dependence of methyl acetate synthesis rate (per g Al; 473 K, 166 kPa CO, 100 kPa DME) on number of H+ in 8-MR of H-FER zeolites synthesized using varying organic SDA (FER-Pyr, FER-TMA, FER-HMI-TMA; Section 2.1.1) or obtained from a commercial source (FER-C). Adapted with permission from Román-Leshkov et al.118 Copyright 2011 American Chemical Society. Dashed line serves to guide the eye. | ||
Using transition state theory,277,278kAc is further described in terms of the Gibbs free energy barrier (ΔGact,Ac) to form the kinetically relevant CO methylation transition state from a surface methyl and a CO molecule in the extracrystalline gas phase (eqn (2); Fig. 11):39
 | (2) |
 | ||
| Fig. 11 Reaction coordinate diagram for CO methylation by a surface methyl species confined within zeolite voids. Adapted from Gounder et al.39 | ||
These studies highlight how kinetically controlled experiments alongside appropriately chosen theoretical models help to establish the identity, number and catalytic consequences of distinct H+ ensembles and further motivate strategies to manipulate the active site environment for desired catalytic transformations. Such catalyst design strategies are enabled by synthetic placement of H+ sites in desired voids via varying organic SDA structures (Section 2.1.1; shown in Fig. 10C for FER) or post-synthetic removal of H+ sites in undesired voids via selective Na+ exchange (Section 2.1.2; shown in Fig. 10B for MOR). Next, we show how the intrinsic selectivity among competing transition states sharing the same precursor state is biased by altering the active site environment for a fixed zeolite using synthetic protocols.
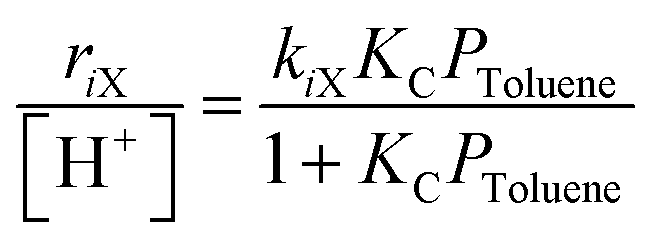 | (3) |
 | ||
| Fig. 12 (A) Total xylene formation rates as a function of toluene pressures during toluene methylation (403 K) by DME (66 kPa) on MFI zeolite samples (Si/Al ∼ 50) synthesized using different organic SDA MFI-DABCO (◆), MFI-EDA (▲) and on a commercial MFI sample surmised to be synthesized using TPA (MFI-TPA-C (●)). Solid lines represent regressed best fits to eqn (3). (B) Average measured xylene isomer selectivity and (C) total xylene formation rate constant as a function of largest cavity diameter on aluminosilicates. Lines represent visual trends for non-MFI aluminosilicates (TON, BEA, MCM-41; ■) and serve to guide the eye. Adapted with permission from Ezenwa et al.131 Copyright 2024 American Chemical Society. | ||
Here, kiX reflects the Gibbs free energy barrier ΔGact,iX for the C–C bond formation transition state relative to the co-adsorbed toluene-surface methyl precursor state (Fig. 13A) and accounts for the number of degenerate ring positions that form each xylene isomer (nC–C,i):131
 | (4) |
 | (5) |
 | ||
| Fig. 13 (A) Gibbs free energy versus reaction coordinate diagram for toluene methylation to xylene on a confined Brønsted acid site showing that kinetic (kiX) and thermodynamic (KC) constants reflect the free energy barriers to form the toluene methylation transition states from co-adsorbed precursor states (ΔGact,iX) and the free energy difference between the co-adsorbed toluene-methyl intermediate and the surface methyl intermediate with toluene in the gas phase, respectively. Free energy differences between individual xylene formation transition states (ΔΔGiX–jX) are independent of the precursor state. Adapted with permission from Ezenwa et al.131 Copyright 2024 American Chemical society (B) DFT-calculated ΔΔGpX–oX for toluene methylation by surface methyls at all 48 T–O pairs in MFI organized by whether the O atoms are accessible to intersection or only accessible to straight or sinusoidal channels. Free energies are reported at 403 K, 1 bar. Reproduced with permission from Ezenwa et al.131 Copyright 2024 American Chemical society. | ||
Relative DFT-calculated barriers for toluene methylation by surface methyls located at all 48 distinct Of atoms show that MFI intersection environments similarly stabilize all three xylene formation transition states (average ΔΔGpX–oX,DFT = 3 kJ mol−1), while channel environments preferentially destabilize the bulkier transition states that form o-X and m-X (average ΔΔGpX–oX,DFT = −22 kJ mol−1) (Fig. 13B).131 Furthermore, enthalpic contributions dominate these Gibbs free energy differences at low temperatures (403 K),131 consistent with the functional form of the equation for Gibbs free energy where entropic losses upon confinement are less dominant.265 Further kinetic and DFT analyses of additional microporous and mesoporous aluminosilicates containing uniform void sizes reveal that more constrained active site environments (∼0.55 nm diameter) preferentially destabilize bulkier transition states, while more spacious active site environments (∼0.70 nm diameter) similarly stabilize all isomer transition states relative to unconfined environments (∼3.0 nm diameter) (Fig. 12B and C).131
These results highlight how the active site environment alters kinetically controlled reaction outcomes when competing transition states have different sizes but share the same precursor state. These insights are further enabled by synthetic techniques that place active sites within distinct confining voids of MFI zeolites. Such active site design strategies complement design strategies that deactivate active sites in unselective pore environments in MWW zeolites for toluene methylation (623 K)30,31 and crystallite design approaches that promote the preferential sieving of the faster diffusing p-X isomer.29,293–295 Furthermore, this active site design approach adds to the growing toolbox of approaches298–301 that aim to design zeolites that contain active site scaffolds which stabilize desired transition states and reactive intermediates for a given reaction.302
3. Active sites at external crystallite surfaces or mesopore environments
In Section 2, we discussed synthesis–structure–reactivity relationships for location of Al (and associated H+) within distinct confining environments (0.4–2.0 nm diameter) of internal micropores. Here in Section 3, we focus on the location of Al (and H+) at unconfined environments (>2.0 nm diameter) of external crystallite surfaces or mesopores that are directly connected to external surfaces (Fig. 14). Our discussion excludes H+ sites that are located within mesopores accessible only from intracrystalline micropore regions (Fig. 14) as such sites possess the limited accessibility of internal sites that excludes certain bulky molecules and the reactivity of external sites with environments that are unable to confine certain reactive intermediates and transition states. We examine synthetic and post-synthetic strategies to control the number of external acid sites in zeolite and present experimental and computational approaches to characterize external acid sites. We further briefly mention a few examples where external active sites have been proposed to influence catalysis. Although we highlight various zeolites, we focus more of the examples in this section on MFI zeolites which are used widely in industrial catalytic applications and are often representative of the broader family of medium pore zeolites possessing limiting apertures that hinder the accessibility of bulk molecules to internal H+ sites. | ||
| Fig. 14 Schematic depicting location of H+ sites at external surfaces and mesopores that are directly connected to external surfaces. H+ sites located within mesopores that are only accessible from internal micropore regions are shown in green. | ||
3.1. Synthetic and post-synthetic strategies to manipulate the number of external acid sites
The number of external H+ sites on a zeolite depends on various crystallite-scale properties including the crystallite size and morphology, the bulk H+ content, and the spatial Al concentration (also known as zoning). Synthetic or post-synthetic techniques that alter these bulk properties, without unintended effects to the number or distribution of internal H+ sites, offer strategies to independently tune the number of external H+ sites and the internal H+ distribution (Fig. 15). | ||
| Fig. 15 Summary of highlighted synthetic and post-synthetic techniques to control the number of external H+ sites. | ||
Synthetic and post-synthetic approaches to tune the crystallite size, morphology, and architecture have been developed for certain zeolites and fall under the realm of crystal engineering, which manipulates the factors (e.g., temperature, pH, identity and concentration of inorganic and organic precursors, seeds, growth modifiers) that control the mechanism and rates of crystal nucleation, growth, and dissolution.69,103,107 Such approaches to produce zeolites possessing nanosized morphologies (e.g., nanoparticles, two-dimensional or layered, or surfaces modified with protrusions or fins) or hierarchical architectures (e.g., pillared, mesoporous) have been summarized by comprehensive reviews and perspectives in the past decade62–75 and will not be discussed here. However, modifying the crystal size and morphology alters the diffusion path length and, in some cases, the effective diffusivity of a molecule in a given framework. Nevertheless, tuning the crystallite size and morphology via synthetic or post-synthetic approaches provides an indirect approach to manipulate the number of external H+ sites (relative to total H+ sites) for a given zeolite framework at fixed total H+ content.
3.2. Characterization of active site locations at external surfaces
External H+ sites charge compensate [AlO4/2]− tetrahedra that are structurally similar to those located within microporous voids. Computational studies that correct for systematic artifacts of periodic DFT methods have shown that the ensemble averaged DPE of H+ sites at all isolated Al locations across different zeolite frameworks (MFI, BEA, FER, MOR, CHA, FAU; 1201 ± 11 kJ mol−1) are similar to those of H+ sites at the unconfined surfaces of mesoporous aluminosilicates (Al-MCM-41; 1212 kJ mol−1).7,8 Recent DFT studies for MFI zeolites have further shown that the ensemble averaged DPE of most external H+ sites are similar to those of internal H+ sites, except for the minority of Brønsted acid sites originating from an H2O molecule coordinating to a trigonal planar Al site that substitutes an external silanol.10 Together, these studies indicate that the strength of Brønsted acid sites is independent of confining void environment. However, external H+ sites differ from internal H+ sites in the inability of unconfined surfaces to restrict the access or formation of bulky species and to provide effective van der Waals stabilization of reactive intermediates and transition states. Thus, techniques to quantify the number of external H+ sites on zeolites mostly depend on the use of stoichiometric probes that selectively titrate only external H+ sites or the use of catalytic probes that exclusively react at external H+ sites.Bulk characterization techniques for Al atoms in zeolites (e.g., elemental analysis, 27Al NMR) are unable to distinguish Al atoms located at surface locations versus interior locations of the zeolite crystallite. As a surface sensitive technique, X-ray photoelectron spectroscopy (XPS) probes the relative Si and Al contents of the crystallite surface and provides useful insights into the presence of spatial concentration gradients of Si and Al atoms across the crystallite length scale,304–306,358,374,380 especially when coupled with depth profiling techniques.320,322,341 However, estimating the number of external H+ sites in zeolites via XPS remains qualitative because XPS provides spatially averaged information of the exterior region of the crystallites (up to 10 nm depth) and requires the assumption that all surface Al are in framework positions and are associated with a proton. Furthermore, low XPS signal-to-noise ratios for zeolite samples containing surface Si/Al > 100 further preclude the use of XPS to reliably estimate the number of external acid sites.339 Nevertheless, XPS provides an approach to assess when the number of external H+ sites deviates significantly from that expected from a homogeneous distribution of H+ across the crystallite.
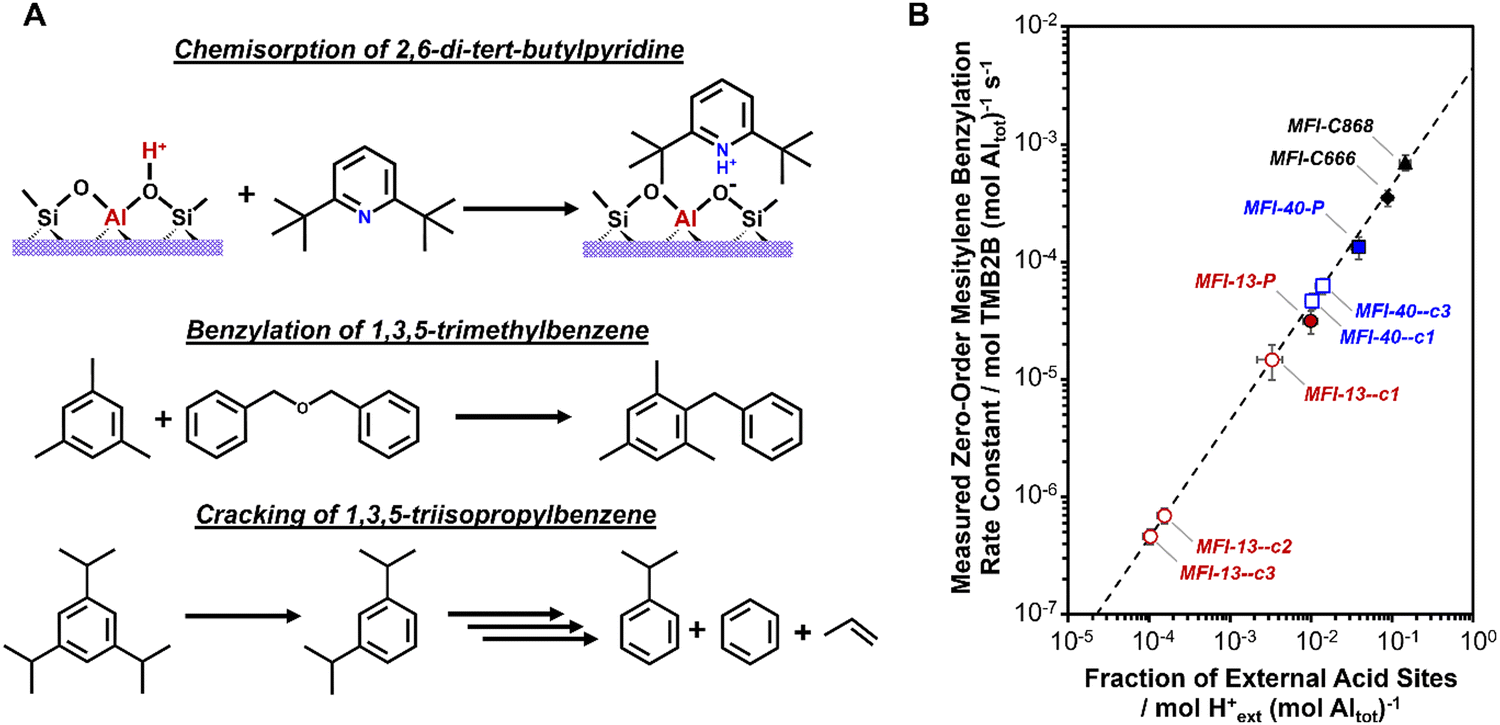
Bulky base molecules (described in Section 3.1.5) are used to selectively titrate external acid sites over internal H+ sites.380 Quantifying the number of external H+ sites requires knowledge of binding stoichiometry (often 1:1 titrant:H+) and that chemisorbed titrants on external Brønsted acid sites can be distinguished from chemisorbed titrants on external Lewis acid sites and physisorbed titrants on external surfaces and mesopores.237,380,383,396 Distinct IR and NMR spectroscopic signatures for chemisorbed titrants on external Brønsted acid sites can be used to distinguish different adsorbed titrants.245,255,384,397–399 Furthermore, alkylpyridine bases containing sterically hindered N centers (e.g., DTBP, TTBP) are unable to coordinate with Lewis acidic Al centers;380,396,400 such features allow certain hindered bases to be used to quantify external H+ sites in situ during steady-state reactions or ex situ using thermogravimetric and temperature-programmed desorption techniques.376,401–403 Together, these highlight how bulky base titrants can be used to quantify the number of external H+ sites in zeolites. However, reliable quantification using stoichiometric probes is limited to samples containing significant amounts of external H+ sites (>10 μmol external H+ g−1), because of typical equipment detection limits.339,376,380,396
Catalytic reactions that react solely on external H+ sites can serve as a probe to quantify the number of external H+ sites in a given zeolite. Various catalytic probe reactions for external H+ sites, mostly in medium-pore zeolites, have been explored over the history of zeolite catalysis and usually involve reactions of bulky aromatic molecules.403,404 The gas-phase cracking of 1,3,5-triisopropylbenzene (dkin ∼ 0.90 nm)334,339,374,380,402 and the liquid-phase benzylation of 1,3,5-trimethylbenzene (mesitylene; dkin ∼ 0.70 nm)381,405–409 are two reactions that are routinely employed in comparative assessments of the number of external H+ sites across medium pore zeolites (typically MFI). Regardless of the choice of a reaction, a quantitative kinetic probe of external H+ sites requires that it solely reflects the concentration of reactive intermediates and kinetically relevant transition states; such requirements can be fulfilled by operating at conditions where measured rates are (i) kinetically controlled without influences of extracrystalline transport restrictions, (ii) in a fixed kinetic regime, preferably first- or zero-order dependence on reactant pressures, (iii) not convoluted by side reactions and deactivation, and (iv) not influenced by approach to equilibrium artifacts. Quantitative kinetic probes for external H+ sites can be further validated using samples containing external H+ sites that can be reliably quantified using a stoichiometric probe (e.g., DTBP titration).381 We demonstrated these in our recent report376 where we developed mesitylene benzylation by dibenzyl ether as a kinetic method to quantify external H+ sites in MFI zeolites obtained after surface passivation treatments (Fig. 16). Catalytic probes, when compared to stoichiometric probes, provide an active site quantification method that amplifies dilute concentrations of external H+ sites, thereby allowing more reliable assessments when the number of external H+ sites is below the detection limits of conventional techniques. Furthermore, these kinetic methods are positioned to advance studies that aim to develop rigorous (post)synthesis–structure–reactivity relationships for external H+ sites during acid catalysis.
 | ||
| Fig. 16 (A) Some stoichiometric and catalytic probes used to assess external H+ sites. (B) Dependence of mesitylene benzylation rates per g on the number of external H+ sites per g zeolite. Measured zero-order rate constants (per total Al) for mesitylene benzylation with DBE (363 K) as a function of fraction of external H+ sites (per total Al) on MFI-13-P (●), MFI-40-P (■), MFI-C666 (◆) and MFI-C868 (▲). The intrinsic mesitylene benzylation rate constant was further used to estimate the fraction of external H+ sites on samples post-synthetically modified with AHFS in multiple treatment cycles (open symbols). Adapted with permission from Ezenwa et al.376 Copyright 2024 Royal Society of Chemistry. | ||
3.3. Catalytic consequences of active sites at external unconfined surfaces
External H+ sites in zeolites have been reported to influence reactivity, selectivity and deactivation behavior during Brønsted acid catalysis, especially under conditions where intracrystalline diffusion constraints restrict access and egress from internal H+ sites. For toluene methylation under kinetically controlled conditions (403 K), external H+ sites exhibit xylene selectivity (o-X ∼ 60%, p-X ∼ 30%, m-X ∼ 10%) similar to that of internal H+ sites at MFI intersections but rates that are ∼102× lower than those of H+ sites at MFI intersections.131 In contrast, earlier toluene methylation studies at higher temperatures (573–773 K) reported that external H+ sites in MFI zeolites alter diffusion controlled selectivity (p-X ∼ 99%) towards thermodynamic equilibrium (o-X ∼ 25%, p-X ∼ 25%, m-X ∼ 50%) by allowing xylene isomerization via bulky transition states that are hindered within micropores.292,334,379,410,411 External H+ sites were also reported to affect oligomer selectivity during propene oligomerization (463–523) in medium pore zeolites (MFI, MTT, TON)359,385,386 and accelerate coke deposition rates on external surfaces during methanol-to-hydrocarbons (743 K) in MFI zeolites.390 Further analysis of the catalytic consequences of external H+ sites is beyond the scope of this perspective.4. Outlook and conclusions: designing active site environments in Brønsted acid zeolites
Catalytic reactions mediated by Brønsted acid zeolites are influenced by the sizes and shapes of the void environments that confine active H+ sites and of the intermediates and transition states stabilized at such sites. In Sections 2 and 3, we presented synthesis–structure–reactivity relationships for active site environments in zeolite catalysis. We discussed how synthetic or post-synthetic approaches can be used to selectively place or remove protons in distinct void environments of given zeolite framework and highlighted characterization strategies that can be used to identify and quantify the location of H+ sites (or framework Al) within distinct void environments (or lattice positions). We further discussed how active site environment influences reactivity and selectivity during catalysis under kinetically controlled conditions. Despite advances in the design of active site environments in zeolites, several challenges remain that present hurdles in our ability to precisely (i) control the location, (ii) identify and quantify the number, and (iii) elucidate the catalytic consequences of active sites in distinct void environments. Here in Section 4, we highlight research opportunities to address knowledge gaps that hinder more precise control and understanding of active site environments in Brønsted acid zeolite catalysis.4.1. Controlling the location of active sites within distinct environments
The framework Al siting in a given zeolite depends on the relative rates and stabilities of Al incorporation at various lattice positions, which in turn depend on the crystallization conditions such as identity and quantity of Si or Al precursors and the SDAs.46,87 Thus, efforts to understand the various interactions between SDAs and the inorganic zeolite framework will aid in clarifying the factors influencing the location and orientation of SDAs within distinct voids features.119,149,171,412–422 The ability to control the location of active sites within distinct void environments will benefit from efforts to deconvolute the effects of structure and mobility of organic SDAs on their interactions with framework Al centers.171,172,419 Even after completion of bulk crystallization, framework Al atoms can be redistributed because of reversible cleavage and formation of Si–O–Al bonds; such Al rearrangements are affected by various factors including crystallization times, temperatures, and SDAs.423–426 Delineating how the temporal Al distribution depends on kinetic control (i.e., dictated by relative rates of Al incorporation among various T-sites) versus thermodynamic control (i.e., dictated by relative stabilities of Al incorporated at various T-sites) remains challenging,87 but is important to further clarify when and how organic SDAs bias the siting of Al centers at various lattice positions.Although the exclusion of bulky base titrants from protons in smaller voids of certain zeolite frameworks can be explained using size-exclusion arguments,174,185–187 the ability of smaller cations (e.g., Na+) to preferentially exchange certain proton ensembles,11,13,182–185 despite their accessibility to all distinct void environments, requires further insights into the intermolecular interactions that dictate such behavior. Molecular-level descriptions of how active sites in distinct confining environments30,192,193,195,198 or at unconfined zeolite surfaces52,332,375,389–391,427 deactivate during catalysis will enable strategies to harness the selective deposition of unreactive residues in tuning active site environments in zeolites. Further insights are needed into the effects of post-synthetic treatment conditions on selective removal of Al atoms from certain T-sites or from external crystallite surfaces373,428,429 and on the selective re-insertion of Al atoms into certain vacancy defects following the removal of relatively unreactive heteroatoms (e.g., B). These efforts to develop deeper insights into the effects of post-synthetic treatments on the selective removal of active sites in undesired void environments will complement the efforts to elucidate the effects of synthetic parameters on the selective placement of active sites in desired environments.
4.2. Identifying and quantifying active sites in distinct void environments
Further advances in 27Al NMR techniques180,220,226,227,430 may enable researchers to more precisely and unambiguously resolve spectroscopic signals for certain zeolite frameworks (e.g., MFI, MWW) into their contributions from distinct framework Al lattice positions. Identifying the highest probability locations of Al atoms in zeolites will also benefit from further advances in XRD techniques119,120,124,230,232 and multinuclear and multidimensional NMR techniques179,180,220,416,431 that probe the location and orientation of organic SDA molecules within voids and their interaction with framework Al centers. Studies to identify probe molecules with distinct spectroscopic (e.g., NMR, IR) signatures when adsorbed at protons or cations within different zeolite void environments could also offer an approach to quantify distinct H+ environments. Emerging techniques such as integrated differential phase contrast scanning transmission electron microscopy (iDPC-STEM)432–435 allow imaging distinct atomic positions of heteroatoms substituted within zeolite frameworks and the orientations of base titrants (e.g., pyridine) within zeolite pores. Substituting lighter N atoms (Z = 7) in base titrants with heavier atoms such as S (Z = 16)433 and framework Al atoms (Z = 13) with heavier heteroatoms such as Ti (Z = 22) or In (Z = 49) may make it possible to use iDPC-STEM, to better resolve atomic positions in the organic guests and zeolite hosts, respectively. Ultimately, advancements in the identification and quantification of distinct Al (and H+) environments will bolster efforts to control the active site environments and to distinguish the intrinsic reactivity of active sites within distinct environments.4.3. Elucidating effects of active site environments on catalysis
Clarifying the effects of active site environments on catalysis requires maintaining the link between reactivity and local environment of active sites and bound species;38,39 this requires that effects of thermodynamics, transport, and deactivation, often present in experimentally measured data, are eliminated or accounted for in the development of kinetic models and in mechanistic interpretations.16,18,19,27,77,131,268,436–438 Thus, efforts to vary the active site environment at fixed bulk zeolite property (e.g., crystallite sizes, active site density) and vice versa,75,200,436,439 and to measure intrinsic diffusional properties (e.g., D/R2)268,436,440,441 may permit the deconvolution of transport effects from intrinsic kinetics at distinct active site environments. When possible, the contributions of external H+ sites to observed reactivity and selectivity should be distinguished from those of internal H+ sites; these contributions depend on the number and reactivity of external H+ sites (relative to internal H+ sites).10,303 Furthermore, techniques to quantify active site ensembles via spectroscopic or titrimetric methods during catalysis are preferable over ex situ techniques that may overestimate the number of catalytically active protons; such techniques also facilitate rigorous assignments of differences in measured turnover rates to the free energy differences between kinetically relevant transition states and reactive intermediates.44,257,442,443The reactions of hydrocarbons (e.g., alkene oligomerization and arene methylation) and oxygenates (e.g., methanol/DME to hydrocarbons) in zeolites often involve complex reaction networks containing ∼102–104 constituent reactions.16,444–446 The nature of such complex reactions create challenges in elucidating effects of active site environments on catalytic reactivity and selectivity. Where possible, isolated constituent reactions or reaction conditions such as lower temperature and conversions that simplify complex networks (<101 reactions) allow more facile connections between measured kinetic and thermodynamic constants to the energetics of specific elementary steps in reaction mechanisms. Continued endeavors to explore catalytic reactions that are sensitive to active site environments, including reaction chemistries of industrial relevance, will enable developing quantitative catalytic probes for distinct active site environments.
Catalysis remains the ultimate arbitrer of differences in active site environments, because under kinetically controlled conditions, reactivity and selectivity differences reflect ensemble averaged reactive encounters with active sites. Thus, the aforementioned experimental efforts to control and quantify active site environments and elucidate their effects on catalysis will be supported by the ensuing theoretical efforts to develop molecular insights into synthesis–structure–reactivity relationships for active site environments in Brønsted acid zeolites.
4.4. Advancing computational approaches to interrogate the synthetic placement and catalytic consequences of distinct Al locations and H+ environments
The vast compositional space encountered during zeolite synthesis will benefit from data-driven approaches to catalogue synthesis information from literature, analyze results from high-throughput experimentations and simulations, and decompose complex synthesis phenomena into tractable descriptors that capture how synthesis parameters regulate Al siting.419,447–456 Such approaches also permit the rapid screening of large numbers of known or hypothetical organic SDAs against all Al locations in a given zeolite to identify promising candidates for further studies using more accurate theoretical simulations.457 The design of zeolites that contain active site environments which stabilize desired transition states and reactive intermediates of a given reaction offers an approach to directly link synthesis to reactivity.298–302 This approach is aided by theoretical calculations and computational screening workflows that assist in identification of organic SDAs that resemble desired transition states or intermediates and candidate zeolites with active environments that stabilize these bound species.298–302 Further advances in designing zeolites via organic SDA mimics may further uncover key features of active site environments that stabilize organic SDAs during synthesis and transition states or intermediates during catalysis.The use of theoretical methods that account for dispersion forces is necessary for describing the effects of active site environment on the stabilities of intermediates and transition states.39,44 In addition, studies that sample the locations of reactive intermediates and transition states across all possible Al and H+ sites provide more insights than those that model only one or two Al lattice positions in lower symmetry frameworks.131 Although such exhaustive approaches are computationally demanding, they are important for comparisons to experiments especially when adjudicating among competing hypotheses on the origins of diverse catalytic behavior for samples whose actual Al distributions are imprecisely known. Macroscopic properties (e.g., kinetic and thermodynamic constants) are typically estimated from statistical averaging of microscopic free energies across all possible Al and H+ locations. However, because the distributions of Al and H+ sites are non-random and often unknown, care should be taken to ensure that theoretical calculations are appropriately used to interpret experimental results. Further advancements in molecular descriptions of catalysis in confined spaces can be achieved by replacing static size descriptors of voids (e.g., PLD, LCD) and bound species (e.g., kinetic diameter,458,459 critical diameter,459–461 van der Waals diameter8,43,268,462) with descriptors such as van der Waals interaction energies that account for the size and shape of non-spherical voids that dynamically restructure to maximize contacts with confined intermediates and transition states.32,43,44,268,463
In summary, further advances in the design of active site environments in zeolites for catalysis will continue to require closely integrated experimental and theoretical approaches to improve fundamental understanding of the molecular-level details that underlie synthesis–structure–reactivity relationships. Zeolites remain one of the most widely used industrial catalysts as Brønsted acids when substituted with trivalent heteroatoms (e.g., Al3+, B3+), as Lewis acids when substituted with electron-deficient tetravalent heteroatoms (e.g., Sn4+, Ti4+, Hf4+), as supports for metal ions (Mn+) and complexes ([MxOy]n+), or in bifunctional formulations with metal nanoparticles and clusters (e.g., Pt). Thus, the ability to precisely place framework heteroatoms in zeolites, especially among the few frameworks (∼10) that have already been scaled up and implemented for commercial applications, will have significant implications for the wide variety of reactions useful in upgrading traditional fossil feedstocks (e.g., crude oil, shale gas) and emerging feedstocks (e.g., biomass, waste plastics, CO2) to higher value chemicals and fuels.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence work reported in this paper.Acknowledgements
We acknowledge many former researchers at Purdue, including Dr Phil Kester, Dr Young Gul Hur, Dr Claire Nimlos, and Dr Byung Jin Lee for their technical and conceptual contributions to the findings described in this feature article and in previously published manuscripts related to controlling the distribution of H+ sites in MFI zeolites. We thank Geoff Hopping, Eric Sauer, Savanna Mack, Teah Scott and Olivia Gluth for their contributions and helpful discussions on various aspects of external H+ sites in MFI zeolites. We acknowledge Prof. David Hibbitts (Univ. of Florida), Dr Alexander Hoffman (Univ. of Florida), Hansel Montalvo-Castro (Univ. of Florida), Dr Deng-Yang Jan (Honeywell UOP), Prof. Bradley Chmelka (Univ. of California Santa Barbara), and Dr Michael Schmithorst (Univ. of California Santa Barbara) for their collaborations and contributions to the findings jointly published and discussed herein. We also acknowledge Andrew Norfleet and Ricem Diaz Arroyo for helpful technical discussions and for careful proofreading of this manuscript. The authors acknowledge financial support provided by the National Science Foundation under Cooperative Agreement No. EEC-1647722, which is an Engineering Research Center for the Innovative and Strategic Transformation of Alkane Resources. S. E. also acknowledges financial support from CISTAR Bill Murray and Dick Reitz Purdue Fellowships.References
- T. F. Degnan, in Studies in Surface Science and Catalysis, ed. R. Xu, Z. Gao, J. Chen and W. Yan, Elsevier, 2007, vol. 170, pp. 54–65 Search PubMed.
- W. Vermeiren and J.-P. Gilson, Top. Catal., 2009, 52, 1131–1161 CrossRef CAS.
- S. I. Zones, Microporous Mesoporous Mater., 2011, 144, 1–8 CrossRef CAS.
- C. Martínez and A. Corma, Coord. Chem. Rev., 2011, 255, 1558–1580 Search PubMed.
- W. O. Haag, R. M. Lago and P. B. Weisz, Nature, 1984, 309, 589 CrossRef CAS.
- W. O. Haag, in Studies in Surface Science and Catalysis, ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Hölderich, Elsevier, 1994, vol. 84, pp. 1375–1394 Search PubMed.
- A. J. Jones and E. Iglesia, ACS Catal., 2015, 5, 5741–5755 Search PubMed.
- S. Wang and E. Iglesia, ACS Catal., 2016, 6, 7664–7684 CrossRef CAS.
- M. Rybicki and J. Sauer, Catal. Today, 2019, 323, 86–93 CrossRef CAS.
- H. Balcom, A. J. Hoffman, H. Locht and D. Hibbitts, ACS Catal., 2023, 13, 4470–4487 CrossRef CAS.
- R. Gounder and E. Iglesia, J. Am. Chem. Soc., 2009, 131, 1958–1971 CrossRef CAS PubMed.
- C. Song, Y. Chu, M. Wang, H. Shi, L. Zhao, X. Guo, W. Yang, J. Shen, N. Xue, L. Peng and W. Ding, J. Catal., 2017, 349, 163–174 CrossRef CAS.
- P. M. Kester, J. T. Crum, S. Li, W. F. Schneider and R. Gounder, J. Catal., 2021, 395, 210–226 CrossRef CAS.
- M. Guisnet and P. Magnoux, Appl. Catal., 1989, 54, 1–27 CrossRef CAS.
- P. M. Kester, E. Iglesia and R. Gounder, J. Phys. Chem. C, 2020, 124, 15839–15855 CrossRef CAS.
- M. L. Sarazen, E. Doskocil and E. Iglesia, ACS Catal., 2016, 6, 7059–7070 CrossRef CAS.
- M. L. Sarazen, E. Doskocil and E. Iglesia, J. Catal., 2016, 344, 553–569 CrossRef CAS.
- E. E. Bickel and R. Gounder, JACS Au, 2022, 2, 2585–2595 CrossRef CAS PubMed.
- E. E. Bickel, S. Lee and R. Gounder, ACS Catal., 2023, 13, 1257–1269 CrossRef CAS.
- R. Gounder and E. Iglesia, J. Catal., 2011, 277, 36–45 CrossRef CAS.
- A. J. Jones, S. I. Zones and E. Iglesia, J. Phys. Chem. C, 2014, 118, 17787–17800 CrossRef CAS.
- J. R. Di Iorio, C. T. Nimlos and R. Gounder, ACS Catal., 2017, 7, 6663–6674 CrossRef CAS.
- A. J. Hoffman, J. S. Bates, J. R. D. Iorio, S. V. Nystrom, C. T. Nimlos, R. Gounder and D. Hibbitts, Angew. Chem., Int. Ed., 2020, 59, 18686–18694 CrossRef CAS PubMed.
- J. S. Bates, B. C. Bukowski, J. Greeley and R. Gounder, Chem. Sci., 2020, 11, 7102–7122 RSC.
- Y. Shen, T. T. Le, D. Fu, J. E. Schmidt, M. Filez, B. M. Weckhuysen and J. D. Rimer, ACS Catal., 2018, 8, 11042–11053 CrossRef CAS.
- Z. S. B. Sousa, D. V. Cesar, C. A. Henriques and V. T. Da Silva, Catal. Today, 2014, 234, 182–191 CrossRef CAS.
- Z. Shi and A. Bhan, J. Catal., 2023, 421, 198–209 CrossRef CAS.
- J. H. Ahn, R. Kolvenbach, S. S. Al-Khattaf, A. Jentys and J. A. Lercher, ACS Catal., 2013, 3, 817–825 CrossRef CAS.
- J. H. Ahn, R. Kolvenbach, O. Y. Gutiérrez, S. S. Al-Khattaf, A. Jentys and J. A. Lercher, Microporous Mesoporous Mater., 2015, 210, 52–59 CrossRef CAS.
- D. Parmar, S. H. Cha, T. Salavati-fard, A. Agarwal, H. Chiang, S. M. Washburn, J. C. Palmer, L. C. Grabow and J. D. Rimer, J. Am. Chem. Soc., 2022, 144, 7861–7870 CrossRef CAS PubMed.
- D. Parmar, S. H. Cha, C. Huang, H. Chiang, S. Washburn, L. C. Grabow and J. D. Rimer, Catal. Sci. Technol., 2023, 13, 5227–5236 RSC.
- S. Wang and E. Iglesia, J. Catal., 2017, 352, 415–435 CrossRef CAS.
- E. S. Vasiliadou, N. S. Gould and R. F. Lobo, ChemCatChem, 2017, 9, 4417–4425 CrossRef CAS.
- P. B. Weisz, Pure Appl. Chem., 1980, 52, 2091–2103 CAS.
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef CAS.
- J. Weitkamp, S. Ernst and L. Puppe, in Catalysis and Zeolites: Fundamentals and Applications, ed. J. Weitkamp and L. Puppe, Springer, Berlin, Heidelberg, 1999, pp. 327–376 Search PubMed.
- T. F. Degnan, J. Catal., 2003, 216, 32–46 CrossRef CAS.
- A. Bhan and E. Iglesia, Acc. Chem. Res., 2008, 41, 559–567 CrossRef CAS PubMed.
- R. Gounder and E. Iglesia, Chem. Commun., 2013, 49, 3491–3509 RSC.
- E. G. Derouane, J. Catal., 1986, 100, 541–544 CrossRef CAS.
- F. Eder and J. A. Lercher, J. Phys. Chem. B, 1997, 101, 1273–1278 CrossRef CAS.
- S. Wang, I. Agirrezabal-Telleria, A. Bhan, D. Simonetti, K. Takanabe and E. Iglesia, Faraday Discuss., 2017, 197, 9–39 RSC.
- M. L. Sarazen and E. Iglesia, ChemCatChem, 2018, 10, 4028–4037 CrossRef CAS.
- E. Iglesia, Catalysis in Chemistry and Biology, World Scientific, 2018, pp. 148–155 Search PubMed.
- P. Deshlahra and E. Iglesia, Chem. Commun., 2020, 56, 7371–7398 RSC.
- B. C. Knott, C. T. Nimlos, D. J. Robichaud, M. R. Nimlos, S. Kim and R. Gounder, ACS Catal., 2018, 8, 770–784 CrossRef CAS.
- E. E. Bickel, C. T. Nimlos and R. Gounder, J. Catal., 2021, 399, 75–85 CrossRef CAS.
- B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317–321 CrossRef CAS PubMed.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed.
- D. T. Bregante, L. N. Wilcox, C. Liu, C. Paolucci, R. Gounder and D. W. Flaherty, ACS Catal., 2021, 11, 11873–11884 CrossRef CAS.
- R. W. Borry, Y. H. Kim, A. Huffsmith, J. A. Reimer and E. Iglesia, J. Phys. Chem. B, 1999, 103, 5787–5796 CrossRef CAS.
- W. Ding, G. D. Meitzner and E. Iglesia, J. Catal., 2002, 206, 14–22 CrossRef CAS.
- Á. N. Santiago-Colón and R. Gounder, J. Catal., 2024, 430, 115335 CrossRef.
- J. A. Biscardi and E. Iglesia, Catal. Today, 1996, 31, 207–231 CrossRef CAS.
- N. M. Phadke, J. Van der Mynsbrugge, E. Mansoor, A. B. Getsoian, M. Head-Gordon and A. T. Bell, ACS Catal., 2018, 8, 6106–6126 CrossRef CAS.
- Y. Yuan, R. F. Lobo and B. Xu, ACS Catal., 2022, 1775–1783 CrossRef CAS.
- A. Finiels, F. Fajula and V. Hulea, Catal. Sci. Technol., 2014, 4, 2412–2426 RSC.
- R. Joshi, G. Zhang, J. T. Miller and R. Gounder, ACS Catal., 2018, 8, 11407–11422 CrossRef CAS.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- C. Paolucci, J. R. Di Iorio, W. F. Schneider and R. Gounder, Acc. Chem. Res., 2020, 53, 1881–1892 CrossRef CAS PubMed.
- V. Valtchev, G. Majano, S. Mintova and J. Pérez-Ramírez, Chem. Soc. Rev., 2013, 42, 263–290 RSC.
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734–6760 CrossRef CAS PubMed.
- W. J. Roth, P. Nachtigall, R. E. Morris and J. Čejka, Chem. Rev., 2014, 114, 4807–4837 CrossRef CAS PubMed.
- J. D. Rimer, M. Kumar, R. Li, A. I. Lupulescu and M. D. Oleksiak, Catal. Sci. Technol., 2014, 4, 3762–3771 RSC.
- Z. Qin, J.-P. Gilson and V. Valtchev, Curr. Opin. Chem. Eng., 2015, 8, 1–6 CrossRef.
- K. N. Olafson, R. Li, B. G. Alamani and J. D. Rimer, Chem. Mater., 2016, 28, 8453–8465 CrossRef CAS.
- N. Masoumifard, R. Guillet-Nicolas and F. Kleitz, Adv. Mater., 2018, 30, 1704439 CrossRef PubMed.
- J. Přech, P. Pizarro, D. P. Serrano and J. Čejka, Chem. Soc. Rev., 2018, 47, 8263–8306 RSC.
- J. D. Rimer, A. Chawla and T. T. Le, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 283–309 CrossRef PubMed.
- R. Bai, Y. Song, Y. Li and J. Yu, Trends Chem., 2019, 1, 601–611 CrossRef CAS.
- S. Li, J. Li, M. Dong, S. Fan, T. Zhao, J. Wang and W. Fan, Chem. Soc. Rev., 2019, 48, 885–907 RSC.
- D. Zhang, C. Jin, M. Zou and S. Huang, Chem. – Eur. J., 2019, 25, 2675–2683 CrossRef CAS PubMed.
- L.-H. Chen, M.-H. Sun, Z. Wang, W. Yang, Z. Xie and B.-L. Su, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed.
- S. Li, H. Yang, S. Wang, J. Wang, W. Fan and M. Dong, Chem. Commun., 2022, 58, 2041–2054 RSC.
- A. J. Mallette, S. Seo and J. D. Rimer, Nat. Synth., 2022, 1, 521–534 CrossRef.
- I. C. Medeiros-Costa, E. Dib, N. Nesterenko, J.-P. Dath, J.-P. Gilson and S. Mintova, Chem. Soc. Rev., 2021, 50, 11156–11179 RSC.
- G. Noh and M. L. Sarazen, J. Catal., 2021, 404, 679–686 CrossRef CAS.
- S. Lee, Y. Park and M. Choi, ACS Catal., 2024, 2031–2048 CrossRef CAS.
- J. Dědeček, Z. Sobalík and B. Wichterlová, Catal. Rev., 2012, 54, 135–223 CrossRef.
- J. Dědeček, E. Tabor and S. Sklenak, ChemSusChem, 2019, 12, 556–576 CrossRef PubMed.
- M. Shamzhy, M. Opanasenko, P. Concepción and A. Martínez, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- S. Wang, Y. He, W. Jiao, J. Wang and W. Fan, Curr. Opin. Chem. Eng., 2019, 23, 146–154 CrossRef.
- A. Palčić and V. Valtchev, Appl. Catal., A., 2020, 606, 117795 CrossRef.
- T. T. Le, A. Chawla and J. D. Rimer, J. Catal., 2020, 391, 56–68 CrossRef CAS.
- S. J. Kwak, H. S. Kim, N. Park, M.-J. Park and W. B. Lee, Korean J. Chem. Eng., 2021, 38, 1117–1128 CrossRef CAS.
- J. Li, M. Gao, W. Yan and J. Yu, Chem. Sci., 2023, 14, 1935–1959 RSC.
- J. Bae and M. Dusselier, Chem. Commun., 2023, 59, 852–867 RSC.
- R. F. Lobo, in Handbook of Zeolite Science and Technology, ed. S. Auerbach, K. Carrado and P. Dutta, Marcel Dekker, Inc., New York, Basel, 2003, pp. 80–112 Search PubMed.
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, https://www.iza-structure.org/databases/, (accessed April 18, 2024).
- M. D. Foster, I. Rivin, M. M. J. Treacy and O. Delgado Friedrichs, Microporous Mesoporous Mater., 2006, 90, 32–38 CrossRef CAS.
- E. Haldoupis, S. Nair and D. S. Sholl, Phys. Chem. Chem. Phys., 2011, 13, 5053–5060 RSC.
- E. L. First, C. E. Gounaris, J. Wei and C. A. Floudas, Phys. Chem. Chem. Phys., 2011, 13, 17339–17358 RSC.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- A. J. Jones, C. Ostrouchov, M. Haranczyk and E. Iglesia, Microporous Mesoporous Mater., 2013, 181, 208–216 CrossRef CAS.
- M. Pinheiro, R. L. Martin, C. H. Rycroft, A. Jones, E. Iglesia and M. Haranczyk, J. Mol. Graphics. Modell., 2013, 44, 208–219 CrossRef CAS PubMed.
- M. Pinheiro, R. L. Martin, C. H. Rycroft and M. Haranczyk, CrystEngComm, 2013, 15, 7531–7538 RSC.
- J. T. Crum, J. R. Crum, C. Taylor and W. F. Schneider, Microporous Mesoporous Mater., 2023, 351, 112466 CrossRef CAS.
- C. E. Webster, R. S. Drago and M. C. Zerner, J. Phys. Chem. B, 1999, 103, 1242–1249 CrossRef CAS.
- C. D. Chang and A. T. Bell, Catal. Lett., 1991, 8, 305–316 CrossRef CAS.
- S. L. Burkett and M. E. Davis, J. Phys. Chem., 1994, 98, 4647–4653 CrossRef CAS.
- S. L. Burkett and M. E. Davis, Chem. Mater., 1995, 7, 920–928 CrossRef CAS.
- S. L. Burkett and M. E. Davis, Chem. Mater., 1995, 7, 1453–1463 CrossRef CAS.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- M. D. Oleksiak, J. A. Soltis, M. T. Conato, R. L. Penn and J. D. Rimer, Chem. Mater., 2016, 28, 4906–4916 CrossRef CAS.
- M. Kumar, M. K. Choudhary and J. D. Rimer, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- D. Parmar, Z. Niu, Y. Liang, H. Dai and J. D. Rimer, Faraday Discuss., 2022, 235, 322–342 RSC.
- A. J. Mallette, K. Shilpa and J. D. Rimer, Chem. Rev., 2024, 124, 3416–3493 CrossRef CAS PubMed.
- H. Gies and B. Marler, Zeolites, 1992, 12, 42–49 CrossRef CAS.
- D. F. Shantz and R. F. Lobo, Chem. Mater., 1998, 10, 4015–4024 CrossRef CAS.
- D. F. Shantz and R. F. Lobo, J. Am. Chem. Soc., 1998, 120, 2482–2483 CrossRef CAS.
- D. F. Shantz, J. Schmedt auf der Günne, H. Koller and R. F. Lobo, J. Am. Chem. Soc., 2000, 122, 6659–6663 CrossRef CAS.
- M. B. Park, D. Jo, H. C. Jeon, C. P. Nicholas, G. J. Lewis and S. B. Hong, Chem. Mater., 2014, 11 Search PubMed.
- D. F. Shantz, C. Fild, H. Koller and R. F. Lobo, J. Phys. Chem. B, 1999, 103, 10858–10865 CrossRef CAS.
- D. F. Shantz, R. F. Lobo, C. Fild and H. Koller, in Studies in Surface Science and Catalysis, ed. A. Corma, F. V. Melo, S. Mendioroz and J. L. G. Fierro, Elsevier, 2000, vol. 130, pp. 845–850 Search PubMed.
- M. J. Sabater and G. Sastre, Chem. Mater., 2001, 13, 4520–4526 CrossRef CAS.
- G. Sastre, V. Fornes and A. Corma, J. Phys. Chem. B, 2002, 106, 701–708 CrossRef CAS.
- A. B. Pinar, C. Márquez-Álvarez, M. Grande-Casas and J. Pérez-Pariente, J. Catal., 2009, 263, 258–265 CrossRef CAS.
- Y. Román-Leshkov, M. Moliner and M. E. Davis, J. Phys. Chem. C, 2011, 115, 1096–1102 CrossRef.
- A. B. Pinar, L. Gómez-Hortigüela, L. B. McCusker and J. Pérez-Pariente, Chem. Mater., 2013, 25, 3654–3661 CrossRef CAS.
- A. B. Pinar, P. A. Wright, L. Gómez-Hortigüela and J. Pérez-Pariente, Microporous Mesoporous Mater., 2010, 129, 164–172 CrossRef CAS.
- L. Gómez-Hortigüela, A. B. Pinar, F. Corà and J. Pérez-Pariente, Chem. Commun., 2010, 46, 2073–2075 RSC.
- A. B. Pinar, L. Gomez-Hortiguela and J. Perez-Pariente, Chem. Mater., 2007, 19, 5617–5626 CrossRef.
- A. B. Pinar, R. García, L. Gómez-Hortigüela and J. Pérez-Pariente, Top. Catal., 2010, 53, 1297–1303 CrossRef CAS.
- W. Chu, X. Liu, Z. Yang, H. Nakata, X. Tan, X. Liu, L. Xu, P. Guo, X. Li and X. Zhu, Chin. J. Catal., 2021, 42, 2078–2087 CrossRef CAS.
- Y. Guo, S. Wang, R. Geng, P. Wang, S. Li, M. Dong, Z. Qin, J. Wang and W. Fan, iScience, 2023, 26, 107748 CrossRef CAS PubMed.
- A. B. Pinar, P. Rzepka, A. J. Knorpp, L. B. McCusker, C. Baerlocher, T. Huthwelker and J. A. van Bokhoven, J. Am. Chem. Soc., 2021, 143, 17926–17930 CrossRef CAS PubMed.
- G. T. Kokotailo, S. L. Lawton, D. H. Olson and W. M. Meier, Nature, 1978, 272, 437–438 CrossRef CAS.
- D. H. Olson, G. T. Kokotailo, S. L. Lawton and W. M. Meier, J. Phys. Chem., 1981, 85, 2238–2243 CrossRef CAS.
- H. van Koningsveld, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 731–735 CrossRef.
- C. T. Nimlos, A. J. Hoffman, Y. G. Hur, B. J. Lee, J. R. Di Iorio, D. D. Hibbitts and R. Gounder, Chem. Mater., 2020, 32, 9277–9298 CrossRef CAS.
- S. Ezenwa, H. Montalvo-Castro, A. J. Hoffman, H. Locht, J. Attebery, D.-Y. Jan, M. Schmithorst, B. Chmelka, D. Hibbitts and R. Gounder, J. Am. Chem. Soc., 2024, 146, 10666–10678 CrossRef CAS PubMed.
- R. J. Argauer and G. R. Landolt, US Pat., US3702886A, 1972.
- X. Tang, W. Chen, W. Dong, Z. Liu, J. Yuan, H. Xia, X. Yi and A. Zheng, Catal. Today, 2022, 405–406, 101–110 CAS.
- E. Dib, T. Mineva, E. Veron, V. Sarou-Kanian, F. Fayon and B. Alonso, J. Phys. Chem. Lett., 2018, 9, 19–24 CrossRef CAS PubMed.
- T. Yokoi, H. Mochizuki, S. Namba, J. N. Kondo and T. Tatsumi, J. Phys. Chem. C, 2015, 119, 15303–15315 CrossRef CAS.
- T. Yokoi, H. Mochizuki, T. Biligetu, Y. Wang and T. Tatsumi, Chem. Lett., 2017, 46, 798–800 CrossRef CAS.
- T. Biligetu, Y. Wang, T. Nishitoba, R. Otomo, S. Park, H. Mochizuki, J. N. Kondo, T. Tatsumi and T. Yokoi, J. Catal., 2017, 353, 1–10 CrossRef CAS.
- S. Park, T. Biligetu, Y. Wang, T. Nishitoba, J. N. Kondo and T. Yokoi, Catal. Today, 2018, 303, 64–70 CrossRef CAS.
- B. M. Lok, T. R. Cannan and C. A. Messina, Zeolites, 1983, 3, 282–291 CrossRef CAS.
- Z. Gabelica, M. Cavez-Bierman, P. Bodart, A. Gourgue and J. B. Nagy, in Studies in Surface Science and Catalysis, ed. B. Držaj, S. Hočevar and S. Pejovnik, Elsevier, 1985, vol. 24, pp. 55–63 Search PubMed.
- E. W. Valyocsik and L. D. Rollmann, Zeolites, 1985, 5, 123–125 CrossRef CAS.
- F. J. Van Der Gaag, J. C. Jansen and H. Van Bekkum, Appl. Catal., 1985, 17, 261–271 CrossRef CAS.
- A. Araya and B. M. Lowe, Zeolites, 1986, 6, 111–118 CrossRef CAS.
- In Studies in Surface Science and Catalysis, ed. P. A. Jacobs and J. A. Martens, Elsevier, 1987, vol. 33, pp. 147–166 Search PubMed.
- S. Schwarz, M. Kojima and C. T. O’Connor, Appl. Catal., 1991, 73, 313–330 CrossRef CAS.
- L. D. Rollmann, J. L. Schlenker, S. L. Lawton, C. L. Kennedy, G. J. Kennedy and D. J. Doren, J. Phys. Chem. B, 1999, 103, 7175–7183 CrossRef CAS.
- L. D. Rollmann, J. L. Schlenker, C. L. Kennedy, G. J. Kennedy and D. J. Doren, J. Phys. Chem. B, 2000, 104, 721–726 CrossRef CAS.
- S. Zanardi, A. Alberti, R. Millini, G. Bellussi and G. Perego, in Studies in Surface Science and Catalysis, ed. R. Aiello, G. Giordano and F. Testa, Elsevier, 2002, vol. 142, pp. 1923–1930 Search PubMed.
- G. Perego, G. Bellussi, R. Millini, A. Alberti and S. Zanardi, Microporous Mesoporous Mater., 2003, 58, 213–223 CrossRef CAS.
- S. Sang, F. Chang, Z. Liu, C. He, Y. He and L. Xu, Catal. Today, 2004, 93–95, 729–734 CrossRef CAS.
- H. Lee, S. I. Zones and M. E. Davis, J. Phys. Chem. B, 2005, 109, 2187–2191 CrossRef CAS PubMed.
- H. Yu, X. Wang and Y. Long, Microporous Mesoporous Mater., 2006, 95, 234–240 CrossRef CAS.
- H. Wang, P. L. Bogdan and R. R. Willis, US Pat., US20110282122A1, 2011.
- S. H. Keoh, W. Chaikittisilp, K. Muraoka, R. R. Mukti, A. Shimojima, P. Kumar, M. Tsapatsis and T. Okubo, Chem. Mater., 2016, 28, 8997–9007 CrossRef CAS.
- L. Meng, B. Mezari, M. G. Goesten and E. J. M. Hensen, Chem. Mater., 2017, 29, 4091–4096 CrossRef CAS PubMed.
- D. Fu, J. E. Schmidt, Z. Ristanović, A. D. Chowdhury, F. Meirer and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2017, 56, 11217–11221 CrossRef CAS PubMed.
- D. Jo and S. B. Hong, Chem. Commun., 2018, 54, 487–490 RSC.
- H. Liu, H. Wang, A.-H. Xing and J.-H. Cheng, J. Phys. Chem. C, 2019, 123, 15637–15647 CrossRef CAS.
- J. Ma, K. Hidaka, M. Ogura and T. Moteki, Cryst. Growth Des., 2023, 23, 8499–8508 CrossRef CAS.
- S. Ren, F. Yang, C. Tian, Y. Yue, W. Zou, W. Hua and Z. Gao, Catalysts, 2023, 13, 1295 CrossRef CAS.
- Y. Wang, X. He, F. Yang, Z. Su and X. Zhu, Ind. Eng. Chem. Res., 2020, 59, 13420–13427 CrossRef CAS.
- L. Guan, M. Liu, H. Liu, L. Zhang, Y. Zhang, Y. Qin, B. He, Y. Mei and Y. Zu, Fuel, 2023, 353, 129230 CrossRef CAS.
- L. Zhao, P.-P. Xiao, Y. Wang, Y. Lu, T. M. Karim, H. Gies and T. Yokoi, ACS Appl. Mater. Interfaces, 2024, 16(14), 17701–17714 CrossRef CAS PubMed.
- R. Gounder, C. T. Nimlos and B. J. Lee, US Pat., US11851337B2, 2023 Search PubMed.
- J. T. Miller, R. Gounder, F. H. Ribeiro, H.-T. Tseng, P. M. Kester, Y. G. Hur and Y. R. Cho, US Pat., US11358912B2, 2022.
- K. Morgan, G. Gainsford and N. Milestone, J. Chem. Soc., Chem. Commun., 1995, 425–426 RSC.
- Y. Wang, J. Yu, Y. Li, Z. Shi and R. Xu, Chem. – Eur. J., 2003, 9, 5048–5055 CrossRef CAS PubMed.
- L. Gómez-Hortigüela, T. Álvaro-Muñoz, B. Bernardo-Maestro and J. Pérez-Pariente, Phys. Chem. Chem. Phys., 2014, 17, 348–357 RSC.
- B. Bernardo-Maestro, F. López-Arbeloa, J. Pérez-Pariente and L. Gómez-Hortigüela, J. Phys. Chem. C, 2015, 119, 28214–28225 CrossRef CAS.
- L. Gómez-Hortigüela and B. Bernardo-Maestro, in Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, ed. L. Gómez-Hortigüela, Springer International Publishing, Cham, 2017, vol. 175, pp. 201–244 Search PubMed.
- L. Gómez-Hortigüela and M. Á. Camblor, in Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials, ed. L. Gómez-Hortigüela, Springer International Publishing, Cham, 2018, pp. 1–41 Search PubMed.
- B. Bernardo-Maestro, J. Li, J. Pérez-Pariente, F. López-Arbeloa and L. Gómez-Hortigüela, Chem. – Eur. J., 2022, 28, e202200702 CrossRef CAS PubMed.
- H. Jongkind, K. P. Datema, S. Nabuurs, A. Seive and W. H. J. Stork, Microporous Mater., 1997, 10, 149–161 CrossRef CAS.
- M. Wang, S. Huang, J. Lü, Z. Cheng, Y. Li, S. Wang and X. Ma, Chin. J. Catal., 2016, 37, 1530–1537 CrossRef CAS.
- Y. Li, M. Yu, K. Cai, M. Wang, J. Lv, R. F. Howe, S. Huang and X. Ma, Phys. Chem. Chem. Phys., 2020, 22, 11374–11381 RSC.
- F. Chen, X.-B. Feng, L.-Y. Zhang, J.-P. Zhao, Z.-M. He, F.-J. Yi, X.-Y. Zhao and J.-P. Cao, Chem. Eng. Sci., 2022, 263, 118110 CrossRef CAS.
- L. Wang, H. Pan, J. Qian, K. Yan, X. Yang, L. Liu, G. Song and H. Zhu, Microporous Mesoporous Mater., 2023, 354, 112569 CrossRef CAS.
- M. Liu, T. Yokoi, M. Yoshioka, H. Imai, J. N. Kondo and T. Tatsumi, Phys. Chem. Chem. Phys., 2014, 16, 4155–4164 RSC.
- Z. Wang, W. Chu, Z. Zhao, Z. Liu, H. Chen, D. Xiao, K. Gong, F. Li, X. Li and G. Hou, J. Phys. Chem. Lett., 2021, 12, 9398–9406 CrossRef CAS PubMed.
- Z. J. Berkson, M.-F. Hsieh, S. Smeets, D. Gajan, A. Lund, A. Lesage, D. Xie, S. I. Zones, L. B. McCusker, C. Baerlocher and B. F. Chmelka, Angew. Chem., Int. Ed., 2019, 58, 6255–6259 CrossRef CAS PubMed.
- K. Muraoka, W. Chaikittisilp, Y. Yanaba, T. Yoshikawa and T. Okubo, Angew. Chem., Int. Ed., 2018, 57, 3742–3746 CrossRef CAS PubMed.
- J. Datka, B. Gil and A. Kubacka, Zeolites, 1997, 18, 245–249 CrossRef CAS.
- M. A. Makarova, A. E. Wilson, B. J. van Liemt, C. M. A. M. Mesters, A. W. de Winter and C. Williams, J. Catal., 1997, 172, 170–177 CrossRef CAS.
- V. A. Veefkind, M. L. Smidt and J. A. Lercher, Appl. Catal., A, 2000, 194–195, 319–332 CrossRef CAS.
- A. Bhan, A. D. Allian, G. J. Sunley, D. J. Law and E. Iglesia, J. Am. Chem. Soc., 2007, 129, 4919–4924 CrossRef CAS PubMed.
- M. Maache, A. Janin, J. C. Lavalley and E. Benazzi, Zeolites, 1995, 15, 507–516 CrossRef CAS.
- N. S. Nesterenko, F. Thibault-Starzyk, V. Montouillout, V. V. Yuschenko, C. Fernandez, J.-P. Gilson, F. Fajula and I. I. Ivanova, Microporous Mesoporous Mater., 2004, 71, 157–166 CrossRef CAS.
- S. Liu, H. Liu, X. Ma, Y. Liu, W. Zhu and Z. Liu, Catal. Sci. Technol., 2020, 10, 4663–4672 RSC.
- R. Liu, S. Zeng, T. Sun, S. Xu, Z. Yu, Y. Wei and Z. Liu, ACS Catal., 2022, 4491–4500 CrossRef CAS.
- J. R. Carpenter, S. Yeh, S. I. Zones and M. E. Davis, J. Catal., 2010, 269, 64–70 CrossRef CAS.
- S. I. Zones and T. V. Harris, Microporous Mesoporous Mater., 2000, 35–36, 31–46 CrossRef CAS.
- E. Besset, D. Meloni, D. Martin, M. Guisnet and L. Schreyeck, in Studies in Surface Science and Catalysis, ed. B. Delmon and G. F. Froment, Elsevier, 1999, vol. 126, pp. 171–178 Search PubMed.
- D. Meloni, D. Martin and M. Guisnet, Appl. Catal., A, 2001, 215, 67–79 CrossRef CAS.
- P. Matias, J. M. Lopes, S. Laforge, P. Magnoux, M. Guisnet and F. Ramôa Ribeiro, Appl. Catal., A, 2008, 351, 174–183 CrossRef CAS.
- S. Laforge, D. Martin, J. L. Paillaud and M. Guisnet, J. Catal., 2003, 220, 92–103 CrossRef CAS.
- S. Laforge, D. Martin and M. Guisnet, Microporous Mesoporous Mater., 2004, 67, 235–244 CrossRef CAS.
- J. Chen, T. Liang, J. Li, S. Wang, Z. Qin, P. Wang, L. Huang, W. Fan and J. Wang, ACS Catal., 2016, 6, 2299–2313 CrossRef CAS.
- T. Liang, J. Chen, S. Wang, P. Wang, Z. Qin, F. Jin, M. Dong, J. Wang and W. Fan, Catal. Sci. Technol., 2022, 12, 6268–6284 RSC.
- Y. Ding, D. Miao, Z. Wang, J. Feng, P. Zhang, R. Yu, X. Cao, X. Pan and X. Bao, ACS Catal., 2023, 13, 14277–14284 CrossRef CAS.
- P. M. Kester, J. T. Miller and R. Gounder, Ind. Eng. Chem. Res., 2018, 57, 6673–6683 CrossRef CAS.
- C. T.-W. Chu, G. H. Kuehl, R. M. Lago and C. D. Chang, J. Catal., 1985, 93, 451–458 CrossRef CAS.
- Q. Zhu, J. N. Kondo, T. Yokoi, T. Setoyama, M. Yamaguchi, T. Takewaki, K. Domen and T. Tatsumi, Phys. Chem. Chem. Phys., 2011, 13, 14598–14605 RSC.
- A. J. Jones, R. T. Carr, S. I. Zones and E. Iglesia, J. Catal., 2014, 312, 58–68 CrossRef CAS.
- S. I. Zones, L. T. Yuen, Y. Nakagawa, R. A. Van NORDSTRAND and S. D. Toto, in Proceedings from the Ninth International Zeolite Conference, ed. R. von Ballmoos, J. B. Higgins and M. M. J. Treacy, Butterworth-Heinemann, 1993, pp. 163–170 Search PubMed.
- C. Y. Chen and S. I. Zones, in Studies in Surface Science and Catalysis, ed. A. Galarneau, F. Fajula, F. Di Renzo and J. Vedrine, Elsevier, 2001, vol. 135, p. 211 Search PubMed.
- H. Koller, C.-Y. Chen and S. I. Zones, Top. Catal., 2015, 58, 451–479 CrossRef CAS.
- S. I. Zones, A. Benin, S.-J. Hwang, D. Xie, S. Elomari and M.-F. Hsieh, J. Am. Chem. Soc., 2014, 136, 1462–1471 CrossRef CAS PubMed.
- C. Schroeder, C. M. Lew, S. I. Zones and H. Koller, Chem. Mater., 2022, 34, 3479–3488 CrossRef CAS.
- S. I. Zones, C. Y. Chen, A. Benin and S.-J. Hwang, J. Catal., 2013, 308, 213–225 CrossRef CAS.
- S. I. Zones, C. Y. Chen, A. Corma, M. T. Cheng, C. L. Kibby, I. Y. Chan and A. W. Burton, J. Catal., 2007, 250, 41–54 CrossRef CAS.
- S. I. Zones, J. Ruan, S. Elomari, O. Terasaki, C. Y. Chen and A. Corma, Solid State Sci., 2011, 13, 706–713 CrossRef CAS.
- J. C. Vega-Vila, J. W. Harris and R. Gounder, J. Catal., 2016, 344, 108–120 CrossRef CAS.
- G. L. Woolery, G. H. Kuehl, H. C. Timken, A. W. Chester and J. C. Vartuli, Zeolites, 1997, 19, 288–296 CrossRef CAS.
- E. Lippmaa, A. Samoson and M. Magi, J. Am. Chem. Soc., 1986, 108, 1730–1735 CrossRef CAS.
- O. H. Han, C.-S. Kim and S. B. Hong, Angew. Chem., Int. Ed., 2002, 41, 469–472 CrossRef CAS PubMed.
- S. Sklenak, J. Dědeček, C. Li, B. Wichterlová, V. Gábová, M. Sierka and J. Sauer, Angew. Chem., Int. Ed., 2007, 46, 7286–7289 CrossRef CAS PubMed.
- J. Dědeček, S. Sklenak, C. Li, B. Wichterlová, V. Gábová, J. Brus, M. Sierka and J. Sauer, in Studies in Surface Science and Catalysis, ed. A. Gédéon, P. Massiani and F. Babonneau, Elsevier, 2008, vol. 174, pp. 781–786 Search PubMed.
- S. Sklenak, J. Dědeček, C. Li, B. Wichterlová, V. Gábová, M. Sierka and J. Sauer, Phys. Chem. Chem. Phys., 2009, 11, 1237–1247 RSC.
- J. Holzinger, P. Beato, L. F. Lundegaard and J. Skibsted, J. Phys. Chem. C, 2018, 122, 15595–15613 CrossRef CAS.
- S. Al-Nahari, E. Dib, C. Cammarano, E. Saint-Germes, D. Massiot, V. Sarou-Kanian and B. Alonso, Angew. Chem., Int. Ed., 2023, 62, e202217992 CrossRef CAS PubMed.
- Y. Zhou, Y. Mu, M.-F. Hsieh, B. Kabius, C. Pacheco, C. Bator, R. M. Rioux and J. D. Rimer, J. Am. Chem. Soc., 2020, 142, 8211–8222 CrossRef CAS PubMed.
- A. Abraham, S.-H. Lee, C.-H. Shin, S. B. Hong, R. Prins and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2004, 6, 3031–3036 RSC.
- A. Vjunov, J. L. Fulton, T. Huthwelker, S. Pin, D. Mei, G. K. Schenter, N. Govind, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2014, 136, 8296–8306 CrossRef CAS PubMed.
- A. Vjunov, J. L. Fulton, T. Huthwelker, S. Pin, D. Mei, G. K. Schenter, N. Govind, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2015, 137, 2409 CrossRef CAS PubMed.
- R. Otomo, T. Nishitoba, R. Osuga, Y. Kunitake, Y. Kamiya, T. Tatsumi and T. Yokoi, J. Phys. Chem. C, 2018, 122, 1180–1191 CrossRef CAS.
- K. Chen, S. Horstmeier, Vy. T. Nguyen, B. Wang, S. P. Crossley, T. Pham, Z. Gan, I. Hung and J. L. White, J. Am. Chem. Soc., 2020, 142, 7514–7523 CrossRef CAS PubMed.
- K. Chen, Z. Gan, S. Horstmeier and J. L. White, J. Am. Chem. Soc., 2021, 143, 6669–6680 CrossRef CAS PubMed.
- A. B. Pinar, R. Verel, J. Pérez-Pariente and J. A. van Bokhoven, Microporous Mesoporous Mater., 2014, 193, 111–114 CrossRef CAS.
- J. Dedecek, M. J. Lucero, C. Li, F. Gao, P. Klein, M. Urbanova, Z. Tvaruzkova, P. Sazama and S. Sklenak, J. Phys. Chem. C, 2011, 115, 11056–11064 CrossRef CAS.
- S. Smeets, L. B. McCusker, C. Baerlocher, S. Elomari, D. Xie and S. I. Zones, J. Am. Chem. Soc., 2016, 138, 7099–7106 CrossRef CAS PubMed.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- S. Smeets, L. B. McCusker, C. Baerlocher, D. Xie, C.-Y. Chen and S. I. Zones, J. Am. Chem. Soc., 2015, 137, 2015–2020 CrossRef CAS PubMed.
- Y. Ma, L. Wang, L. Chen, M. Shen, X. Yang, T. Wang, F. Yuan, Y. Zhou, J. Wang and H. Zhu, CrystEngComm, 2022, 24, 2118–2125 RSC.
- W. I. F. David and K. Shankland, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 52–64 CrossRef CAS PubMed.
- R. Bohinc, J. Hoszowska, J.-C. Dousse, W. Błachucki, F. Zeeshan, Y. Kayser, M. Nachtegaal, A. B. Pinar and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2017, 19, 29271–29277 RSC.
- J. A. van Bokhoven, T.-L. Lee, M. Drakopoulos, C. Lamberti, S. Thieß and J. Zegenhagen, Nat. Mater., 2008, 7, 551–555 CrossRef CAS PubMed.
- K. Hadjiivanov, in Advances in Catalysis, ed. F. C. Jentoft, Academic Press, 2014, vol. 57, pp. 99–318 Search PubMed.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- M. A. Makarova, V. L. Zholobenko, K. M. Al-Ghefaili, N. E. Thompson, J. Dewing and J. Dwyer, J. Chem. Soc., Faraday Trans., 1994, 90, 1047–1054 RSC.
- V. L. Zholobenko, D. B. Lukyanov, J. Dwyer and W. J. Smith, J. Phys. Chem. B, 1998, 102, 2715–2721 CrossRef CAS.
- P. A. Jacobs and W. J. Mortier, Zeolites, 1982, 2, 226–230 CrossRef CAS.
- T. Bucko, L. Benco, T. Demuth and J. Hafner, J. Chem. Phys., 2002, 117, 7295–7305 CrossRef CAS.
- J. A. Lercher and A. Jentys, in Studies in Surface Science and Catalysis, ed. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, 2007, vol. 168, pp. 435–476 Search PubMed.
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC.
- L. Lakiss, A. Vicente, J.-P. Gilson, V. Valtchev, S. Mintova, A. Vimont, R. Bedard, S. Abdo and J. Bricker, ChemPhysChem, 2020, 21, 1873–1881 CrossRef CAS PubMed.
- A. A. Gabrienko, I. G. Danilova, S. S. Arzumanov, L. V. Pirutko, D. Freude and A. G. Stepanov, J. Phys. Chem. C, 2018, 122, 25386–25395 CrossRef CAS.
- P. A. Jacobs and R. Von Ballmoos, J. Phys. Chem., 1982, 86, 3050–3052 CrossRef CAS.
- T. R. Hughes and H. M. White, J. Phys. Chem., 1967, 71, 2192–2201 CrossRef CAS.
- B. Athokpam, S. G. Ramesh and R. H. McKenzie, Chem. Phys., 2017, 488–489, 43–54 CrossRef CAS.
- H. Tsubomura, J. Chem. Phys., 1956, 24, 927–931 CrossRef CAS.
- L. Treps, C. Demaret, D. Wisser, B. Harbuzaru, A. Méthivier, E. Guillon, D. V. Benedis, A. Gomez, T. D. Bruin, M. Rivallan, L. Catita, A. Lesage and C. Chizallet, J. Phys. Chem. C, 2021, 125, 2163–2181 CrossRef CAS.
- R. A. Schoonheydt and J. B. Uytterhoeven, J. Catal., 1970, 19, 55–63 CrossRef CAS.
- F. Göltl, A. M. Love, S. C. Schuenzel, P. Wolf, M. Mavrikakis and I. Hermans, Phys. Chem. Chem. Phys., 2019, 21, 19065–19075 RSC.
- M. Boronat, C. Martínez-Sánchez, D. Law and A. Corma, J. Am. Chem. Soc., 2008, 130, 16316–16323 CrossRef CAS PubMed.
- M. Dyballa, Energy Fuels, 2023, 37(23), 18517–18559 CrossRef CAS.
- J. A. Lercher, C. Grundling and G. Eder-Mirth, Catal. Today, 1996, 27, 353–376 CrossRef CAS.
- C. J. Wrasman, A. T. Bell, B. D. Chandler, J. W. Harris, S. Kwon, M. R. Ball, S. H. Krishna, S. J. Khatib, P. Bollini, Y. Román-Leshkov, A. “Bean” Getsoian, R. S. Weber, J. A. Lercher, D. Liu, D. E. Resasco, J. S. Bates, J. N. Hall, E. A. Lebrón-Rodríguez, L. Paz Herrera, J. M. Notestein and J. A. Schaidle, J. Catal., 2024, 115451 CrossRef CAS.
- V. J. Frillette, W. O. Haag and R. M. Lago, J. Catal., 1981, 67, 218–222 CrossRef CAS.
- Y. Traa, S. Sealy and J. Weitkamp, in Characterization II, ed. H. G. Karge and J. Weitkamp, Springer, Berlin, Heidelberg, 2007, pp. 103–154 Search PubMed.
- M. D. Macedonia and E. J. Maginn, AIChE J., 2000, 46, 2504–2517 CrossRef CAS.
- W. O. Haag, R. M. Dessau and R. M. Lago, in Studies in Surface Science and Catalysis, ed. T. Lnui, S. Namba and T. Tatsumi, Elsevier, 1991, vol. 60, pp. 255–265 Search PubMed.
- W. O. Haag, R. M. Lago and P. B. Weisz, Faraday Discuss. Chem. Soc., 1981, 72, 317–330 RSC.
- M. L. Sarazen and E. Iglesia, J. Catal., 2017, 354, 287–298 CrossRef CAS.
- J. Van der Mynsbrugge, J. De Ridder, K. Hemelsoet, M. Waroquier and V. Van Speybroeck, Chem. – Eur. J., 2013, 19, 11568–11576 CrossRef CAS PubMed.
- R. Gounder and E. Iglesia, Acc. Chem. Res., 2012, 45, 229–238 CrossRef CAS PubMed.
- M. Boudart, Chem. Rev., 1995, 95, 661–666 CrossRef CAS.
- C. W. Hullfish, J. Z. Tan, H. I. Adawi and M. L. Sarazen, ACS Catal., 2023, 13140–13150 CrossRef CAS.
- G. Noh, Z. Shi, S. I. Zones and E. Iglesia, J. Catal., 2018, 368, 389–410 CrossRef CAS.
- S. Wang, L. Zhang, S. Li, Z. Qin, D. Shi, S. He, K. Yuan, P. Wang, T.-S. Zhao, S. Fan, M. Dong, J. Li, W. Fan and J. Wang, J. Catal., 2019, 377, 81–97 CrossRef CAS.
- S. Wang, P. Wang, Z. Qin, Y. Chen, M. Dong, J. Li, K. Zhang, P. Liu, J. Wang and W. Fan, ACS Catal., 2018, 8, 5485–5505 CrossRef CAS.
- T. Liang, J. Chen, Z. Qin, J. Li, P. Wang, S. Wang, G. Wang, M. Dong, W. Fan and J. Wang, ACS Catal., 2016, 6, 7311–7325 CrossRef CAS.
- K. Fujimoto, T. Shikada, K. Omata and H. Tominaga, Chem. Lett., 1984, 2047–2050 CrossRef CAS.
- P. Cheung, E. Iglesia, J. G. Sunley, D. J. Law and A. Bhan, US Pat., US7465822B2, 2008.
- C. Chizallet, C. Bouchy, K. Larmier and G. Pirngruber, Chem. Rev., 2023, 123, 6107–6196 CrossRef CAS PubMed.
- P. Cheung, A. Bhan, G. J. Sunley and E. Iglesia, Angew. Chem., Int. Ed., 2006, 45, 1617–1620 CrossRef CAS PubMed.
- P. Cheung, A. Bhan, G. J. Sunley, D. J. Law and E. Iglesia, J. Catal., 2007, 245, 110–123 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
- M. Boronat, C. Martínez and A. Corma, Phys. Chem. Chem. Phys., 2011, 13, 2603–2612 RSC.
- D. B. Rasmussen, J. M. Christensen, B. Temel, F. Studt, P. G. Moses, J. Rossmeisl, A. Riisager and A. D. Jensen, Catal. Sci. Technol., 2017, 7, 1141–1152 RSC.
- Y. Chu, A.-Y. Lo, C. Wang and F. Deng, J. Phys. Chem. C, 2019, 123, 15503–15512 CrossRef CAS.
- M. Neurock, Ind. Eng. Chem. Res., 2010, 49, 10183–10199 CrossRef CAS.
- M. Gešvandtnerová, D. Rocca and T. Bučko, J. Catal., 2021, 396, 166–178 CrossRef.
- P. Feng, G. Zhang, X. Chen, K. Zang, X. Li and L. Xu, Appl. Catal., A., 2018, 557, 119–124 CrossRef CAS.
- M. Lusardi, T. T. Chen, M. Kale, J. H. Kang, M. Neurock and M. E. Davis, ACS Catal., 2020, 10, 842–851 CrossRef CAS.
- J. Yao, Q. Wu, J. Fan, S. Komiyama, X. Yong, W. Zhang, T. Zhao, Z. Guo, G. Yang and N. Tsubaki, ACS Nano, 2021, 15, 13568–13578 CrossRef CAS PubMed.
- X. Feng, J. Yao, H. Li, Y. Fang, Y. Yoneyama, G. Yang and N. Tsubaki, Chem. Commun., 2019, 55, 1048–1051 RSC.
- Z. Xiong, E. Zhan, M. Li and W. Shen, Chem. Commun., 2020, 56, 3401–3404 RSC.
- X.-B. Feng, J.-P. Cao, C. Su, Z.-M. He and X.-Y. Zhao, Fuel, 2022, 315, 123267 CrossRef CAS.
- R. D. Chirico and W. V. Steele, J. Chem. Eng. Data, 1997, 42, 784–790 CrossRef CAS.
- N. Chakinala and A. G. Chakinala, Ind. Eng. Chem. Res., 2021, 60, 5331–5351 CrossRef CAS.
- D. Fraenkel, Ind. Eng. Chem. Res., 1990, 29, 1814–1821 CrossRef CAS.
- L. B. Young, S. A. Butter and W. W. Kaeding, J. Catal., 1982, 76, 418–432 CrossRef CAS.
- J. Wei, J. Catal., 1982, 76, 433–439 CrossRef CAS.
- G. Mirth and J. A. Lercher, J. Catal., 1994, 147, 199–206 CrossRef CAS.
- J. E. Leffler, Science, 1953, 117, 340–341 CrossRef CAS PubMed.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- E. M. Gallego, M. T. Portilla, C. Paris, A. León-Escamilla, M. Boronat, M. Moliner and A. Corma, Science, 2017, 355, 1051–1054 CrossRef CAS PubMed.
- C. Li, C. Paris, J. Martínez-Triguero, M. Boronat, M. Moliner and A. Corma, Nat. Catal., 2018, 1, 547–554 CrossRef CAS.
- C. Li, P. Ferri, C. Paris, M. Moliner, M. Boronat and A. Corma, J. Am. Chem. Soc., 2021, 143, 10718–10726 CrossRef CAS PubMed.
- P. Ferri, C. Li, D. Schwalbe-Koda, M. Xie, M. Moliner, R. Gómez-Bombarelli, M. Boronat and A. Corma, Nat. Commun., 2023, 14, 2878 CrossRef CAS PubMed.
- M. Moliner and M. Boronat, Microporous Mesoporous Mater., 2023, 358, 112354 CrossRef CAS.
- M. Farcasiu and T. F. Degnan, Ind. Eng. Chem. Res., 1988, 27, 45–47 CrossRef CAS.
- J. P. Gilson and E. G. Derouane, J. Catal., 1984, 88, 538–541 CrossRef CAS.
- J.-Fr Tempere, D. Delafosse and J. P. Contour, Chem. Phys. Lett., 1975, 33, 95–98 CrossRef CAS.
- J.-F. R. Tempere, D. Delafosse and J. P. Contour, in Molecular Sieves—II, American Chemical Society, 1977, vol. 40, pp. 76–85 Search PubMed.
- R. von Ballmoos and W. M. Meier, Nature, 1981, 289, 782 CrossRef CAS.
- E. G. Derouane, S. Determmerie, Z. Gabelica and N. Blom, Appl. Catal., 1981, 1, 201–224 CrossRef CAS.
- E. G. Derouane, J. P. Gilson, Z. Gabelica, C. Mousty-Desbuquoit and J. Verbist, J. Catal., 1981, 71, 447–448 CrossRef CAS.
- A. E. Hughes, K. G. Wilshier, B. A. Sexton and P. Smart, J. Catal., 1983, 80, 221–227 CrossRef CAS.
- K. Chao and J. Chern, Zeolites, 1988, 8, 82–85 CrossRef CAS.
- R. M. Dessau, E. W. Valyocsik and N. H. Goeke, Zeolites, 1992, 12, 776–779 CrossRef CAS.
- R. Althoff, B. Schulz-Dobrick, F. Schüth and K. Unger, Microporous Mater., 1993, 1, 207–218 CrossRef CAS.
- C. S. Cundy, M. S. Henty and R. J. Plaisted, Zeolites, 1995, 15, 342–352 CrossRef CAS.
- S. A. Schunk, D. G. Demuth, B. Schulz-Dobrick, K. K. Unger and F. Schüth, Microporous Mater., 1996, 6, 273–285 CrossRef CAS.
- N. Danilina, F. Krumeich, S. A. Castelanelli and J. A. van Bokhoven, J. Phys. Chem. C, 2010, 114, 6640–6645 CrossRef CAS.
- J. Shin, N. H. Ahn, S. J. Cho, L. Ren, F.-S. Xiao and S. B. Hong, Chem. Commun., 2014, 50, 1956–1958 RSC.
- A. Lombard, A. Simon-Masseron, L. Rouleau, A. Cabiac and J. Patarin, Microporous Mesoporous Mater., 2010, 129, 220–227 CrossRef CAS.
- Z. Ristanović, J. P. Hofmann, U. Deka, T. U. Schülli, M. Rohnke, A. M. Beale and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 13382–13386 CrossRef PubMed.
- S. R. Bare, A. Knop-Gericke, D. Teschner, M. Hävacker, R. Blume, T. Rocha, R. Schlögl, A. S. Y. Chan, N. Blackwell, M. E. Charochak, R. ter Veen and H. H. Brongersma, Surf. Sci., 2016, 648, 376–382 CrossRef CAS.
- A. Chawla, R. Li, R. Jain, R. John Clark, J. G. Sutjianto, J. C. Palmer and J. D. Rimer, Mol. Syst. Des. EngMol. Syst. Des. Eng., 2018, 3, 159–170 RSC.
- C. Wang, L. Zhang, X. Huang, Y. Zhu, G. (Kevin) Li, Q. Gu, J. Chen, L. Ma, X. Li, Q. He, J. Xu, Q. Sun, C. Song, M. Peng, J. Sun and D. Ma, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- T. Li, F. Krumeich, M. Chen, Z. Ma and J. van Bokhoven, Phys. Chem. Chem. Phys., 2020, 22, 734–739 RSC.
- T. Li, F. Krumeich and J. A. van Bokhoven, Cryst. Growth Des., 2019, 19(5), 2548–2551 CrossRef CAS.
- W. Qin, Y. Zhou and J. D. Rimer, React. Chem. Eng., 2019, 4, 1957–1968 RSC.
- Y. Shen, Z. Qin, S. Asahina, N. Asano, G. Zhang, S. Qian, Y. Ma, Z. Yan, X. Liu and S. Mintova, J. Mater. Chem. A, 2021, 9, 4203–4212 RSC.
- T. T. Le, W. Qin, A. Agarwal, N. Nikolopoulos, D. Fu, M. D. Patton, C. Weiland, S. R. Bare, J. C. Palmer, B. M. Weckhuysen and J. D. Rimer, Nat. Catal., 2023, 1–12 Search PubMed.
- Y. Shen, S. Zhang, Z. Qin, A. Beuque, L. Pinard, S. Asahina, N. Asano, R. Zhang, J. Zhao, Y. Fan, X. Liu, Z. Yan and S. Mintova, ACS Catal., 2024, 3766–3777 CrossRef CAS.
- F. Jiao, P. Yu, Y. Cui, H. Li, Q. Hu, Y. Xu, S. Mintova, H. Guo and H. Du, Angew. Chem., Int. Ed., 2023, e202310419 CAS.
- L. D. Rollmann, US Pat., US4088605A, 1978.
- Q. Li, Z. Wang, J. Hedlund, D. Creaser, H. Zhang, X. Zou and A.-J. Bons, Microporous Mesoporous Mater., 2005, 78, 1–10 CrossRef CAS.
- D. Van Vu, M. Miyamoto, N. Nishiyama, Y. Egashira and K. Ueyama, J. Catal., 2006, 243, 389–394 CrossRef.
- M. Okamoto and Y. Osafune, Microporous Mesoporous Mater., 2011, 143, 413–418 CrossRef CAS.
- D. Yi, X. Xu, X. Meng, N. Liu and L. Shi, J. Porous Mater., 2019, 26, 1767–1779 CrossRef CAS.
- A. M. Goossens, B. H. Wouters, V. Buschmann and J. A. Martens, Adv. Mater., 1999, 11, 561–564 CrossRef CAS.
- Y. Bouizi, I. Diaz, L. Rouleau and V. P. Valtchev, Adv. Funct. Mater., 2005, 15, 1955–1960 CrossRef CAS.
- Y. Bouizi, L. Rouleau and V. P. Valtchev, Chem. Mater., 2006, 18, 4959–4966 CrossRef CAS.
- G. D. Pirngruber, C. Laroche, M. Maricar-Pichon, L. Rouleau, Y. Bouizi and V. Valtchev, Microporous Mesoporous Mater., 2013, 169, 212–217 CrossRef CAS.
- A. Ghorbanpour, A. Gumidyala, L. C. Grabow, S. P. Crossley and J. D. Rimer, ACS Nano, 2015, 9, 4006–4016 CrossRef CAS PubMed.
- S. Han, K. Shilpa, A. J. Mallette, Y. Li, J. B. Hoke and J. D. Rimer, J. Cryst. Growth, 2022, 126992 Search PubMed.
- T. T. Le, K. Shilpa, C. Lee, S. Han, C. Weiland, S. R. Bare, P. J. Dauenhauer and J. D. Rimer, J. Catal., 2022, 405, 664–675 CrossRef CAS.
- M. Niwa, M. Kato, T. Hattori and Y. Murakami, J. Phys. Chem., 1986, 90, 6233–6237 CrossRef CAS.
- T. Hibino, M. Niwa and Y. Murakami, Zeolites, 1993, 13, 518–523 CrossRef CAS.
- Y. S. Bhat, J. Das, K. V. Rao and A. B. Halgeri, J. Catal., 1996, 159, 368–374 CrossRef CAS.
- J.-H. Kim, A. Ishida, M. Okajima and M. Niwa, J. Catal., 1996, 161, 387–392 CrossRef CAS.
- C. D. Chang and P. G. Rodewald, US Pat., US5607888A, 1997.
- S. Zheng, H. R. Heydenrych, A. Jentys and J. A. Lercher, J. Phys. Chem. B, 2002, 106, 9552–9558 CrossRef CAS.
- S. Zheng, H. R. Heydenrych, H. P. Röger, A. Jentys and J. A. Lercher, Top. Catal., 2003, 22, 101–106 CrossRef CAS.
- R. Khare, D. Millar and A. Bhan, J. Catal., 2015, 321, 23–31 CrossRef CAS.
- S. Hu, J. Liu, J. Chen, J. Meng, G. Ye and X. Zhou, Ind. Eng. Chem. Res., 2022, 61, 5747–5756 CrossRef CAS.
- Y. Wu, J. Han, W. Zhang, Z. Yu, K. Wang, X. Fang, Y. Wei and Z. Liu, J. Am. Chem. Soc., 2024, 146(12), 8086–8097 CrossRef CAS PubMed.
- A. Corma, V. Fornés, O. Pallota, J. M. Cruz and A. Ayerbe, J. Chem. Soc., Chem. Commun., 1986, 0, 333–334 RSC.
- G. Garralón, V. Fornés and A. Corma, Zeolites, 1988, 8, 268–272 CrossRef.
- B. Chauvin, M. Boulet, P. Massiani, F. Fatula, F. Figueras and T. Des Courieres, J. Catal., 1990, 126, 532–545 CrossRef CAS.
- Q. L. Wang, M. Torrealba, G. Giannetto, M. Guisnet, G. Perot, M. Cahoreau and J. Caisso, Zeolites, 1990, 10, 703–706 CrossRef CAS.
- E. Bowes and D. S. Shihabi, US Pat., US5080878A, 1992.
- E. Benazzi, J. M. Silva, M. F. Ribeiro, F. R. Ribeiro and M. Guisnet, in Studies in Surface Science and Catalysis, ed. L. Bonneviot and S. Kaliaguine, Elsevier, 1995, vol. 97, pp. 393–400 Search PubMed.
- J. M. Silva, M. F. Ribeiro, F. R. Ribeiro, N. S. Gnep, M. Guisnet and E. Benazzi, React. Kinet. Catal. Lett., 1995, 54, 209–215 CrossRef CAS.
- S. Han, D. S. Shihabi and C. D. Chang, J. Catal., 2000, 196, 375–378 CrossRef CAS.
- P. Cañizares and A. Carrero, Appl. Catal., A., 2003, 248, 227–237 CrossRef.
- M. R. Apelian, T. F. Degnan and A. S. Fung, US Pat., US5234872A, 1993.
- H. A. Zaidi and K. K. Pant, Ind. Eng. Chem. Res., 2008, 47, 2970–2975 CrossRef CAS.
- J. Lv, Z. Hua, J. Zhou, Z. Liu, H. Guo and J. Shi, ChemCatChem, 2018, 10, 2278–2284 CrossRef CAS.
- J. Li, L. Wang, D. Zhang, J. Qian, L. Liu and J. Xing, J. Fuel Chem. Technol., 2019, 47, 957–963 CrossRef CAS.
- J. Dwyer, F. R. Fitch, F. Machado, G. Qin, S. M. Smyth and J. C. Vickerman, J. Chem. Soc., Chem. Commun., 1981, 422–424 RSC.
- S. Inagaki, S. Shinoda, Y. Kaneko, K. Takechi, R. Komatsu, Y. Tsuboi, H. Yamazaki, J. N. Kondo and Y. Kubota, ACS Catal., 2013, 3, 74–78 CrossRef CAS.
- R. Feng, X. Yan, X. Hu, Z. Yan, J. Lin, Z. Li, K. Hou and M. J. Rood, Catal. Commun., 2018, 109, 1–5 CrossRef CAS.
- A. Corma, V. Fornés and F. Rey, Appl. Catal., 1990, 59, 267–274 CrossRef CAS.
- R. Gounder, A. J. Jones, R. T. Carr and E. Iglesia, J. Catal., 2012, 286, 214–223 CrossRef CAS.
- S. Schallmoser, T. Ikuno, M. F. Wagenhofer, R. Kolvenbach, G. L. Haller, M. Sanchez-Sanchez and J. A. Lercher, J. Catal., 2014, 316, 93–102 CrossRef CAS.
- K. Lee, S. Lee, Y. Jun and M. Choi, J. Catal., 2017, 347, 222–230 CrossRef CAS.
- M. Abdolrahmani, K. Chen and J. L. White, J. Phys. Chem. C, 2018, 122, 15520–15528 CrossRef CAS.
- K. Chen, A. Zornes, V. Nguyen, B. Wang, Z. Gan, S. P. Crossley and J. L. White, J. Am. Chem. Soc., 2022, 144, 16916–16929 CrossRef CAS PubMed.
- S. Namba, A. Inaka and T. Yashima, Zeolites, 1986, 6, 107–110 CrossRef CAS.
- J. R. Anderson, Y.-F. Chang and R. J. Western, J. Catal., 1989, 118, 466–482 CrossRef CAS.
- S. Ezenwa, G. M. Hopping, E. D. Sauer, T. Scott, S. Mack and R. Gounder, React. Chem. Eng., 2024, 9, 1096–1112 RSC.
- J.-H. Kim, A. Ishida and M. Niwa, React. Kinet. Catal. Lett., 1999, 67, 281–287 CrossRef CAS.
- S. Namba, S. Nakanishi and T. Yashima, J. Catal., 1984, 88, 505–508 CrossRef CAS.
- T. Yashima, Y. Sakaguchi and S. Namba, in Studies in Surface Science and Catalysis, ed. T. Seivama and K. Tanabe, Elsevier, 1981, vol. 7, pp. 739–751 Search PubMed.
- A. Corma, V. Fornés, L. Forni, F. Márquez, J. Martínez-Triguero and D. Moscotti, J. Catal., 1998, 179, 451–458 CrossRef CAS.
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. A. Wahedi, A. Bhan, S. A. Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Science, 2012, 336, 1684–1687 CrossRef CAS PubMed.
- D. Liu, X. Zhang, A. Bhan and M. Tsapatsis, Microporous Mesoporous Mater., 2014, 200, 287–290 CrossRef CAS.
- M. S. Holm, S. Svelle, F. Joensen, P. Beato, C. H. Christensen, S. Bordiga and M. Bjørgen, Appl. Catal., A, 2009, 356, 23–30 CrossRef CAS.
- C. Freitas, N. S. Barrow and V. Zholobenko, Johnson. Matthey Technol. Rev., 2018, 62, 279–290 CrossRef CAS.
- N. M. Page and L. B. Young, US Pat., US4855527A, 1989.
- J. E. Stanat, G. M. K. Mathys, D. W. Turner, J. C. Cheng, S. W. Beadle, C. M. C. Guajardo, R. Eijkhoudt, A. D. Godwin, E. E. Green, C. M. Yarbrough, R. F. Caers, C. B. Duncan and R. Y. Saleh, US Pat., US7425662B2, 2008.
- H. K. Heinichen and W. F. Hölderich, J. Catal., 1999, 185, 408–414 CrossRef CAS.
- T. W. Beutel, A. M. Willard, C. Lee, M. S. Martinez and R. Dugan, J. Phys. Chem. C, 2021, 125(16), 8518–8532 CrossRef CAS.
- A. G. Popov, V. S. Pavlov and I. I. Ivanova, J. Catal., 2016, 335, 155–164 CrossRef CAS.
- Y. Zhai, S. Zhang, Y. Shang, Y. Song, W. Wang, T. Ma, L. Zhang, Y. Gong, J. Xu and F. Deng, Catal. Sci. Technol., 2019, 9, 659–671 RSC.
- Y. Gan, Q. Lv, Y. Li, H. Yang, K. Xu, L. Wu, Y. Tang and L. Tan, Chem. Eng. Sci., 2023, 266, 118289 CrossRef CAS.
- L.-Y. Fang, S.-B. Liu and I. Wang, J. Catal., 1999, 185, 33–42 CrossRef CAS.
- F. Bauer, W. H. Chen, E. Bilz, A. Freyer, V. Sauerland and S. B. Liu, J. Catal., 2007, 251, 258–270 CrossRef CAS.
- M. Molinier and J. S. Abichandani, in Industrial Arene Chemistry, John Wiley & Sons, Ltd, 2023, pp. 357–382 Search PubMed.
- M. Molinier, R. G. Tinger, A. Cotte and J. S. Abichandani, in Industrial Arene Chemistry, John Wiley & Sons, Ltd, 2023, pp. 327–355 Search PubMed.
- K. Góra-Marek, K. Tarach and M. Choi, J. Phys. Chem. C, 2014, 118, 12266–12274 CrossRef.
- C. Rieg, Z. Li, A. Kurtz, M. Schmidt, D. Dittmann, M. Benz and M. Dyballa, J. Phys. Chem. C, 2021, 125(1), 515–525 CrossRef CAS.
- N. S. Nesterenko, F. Thibault-Starzyk, V. Montouilliout, V. V. Yushchenko, C. Fernandez, J.-P. Gilson, F. Fajula and I. I. Ivanova, Kinet. Catal., 2006, 47, 40–48 CrossRef CAS.
- D. Fărcaşiu, R. Leu and A. Corma, J. Phys. Chem. B, 2002, 106, 928–932 CrossRef.
- C. D. Baertsch, K. T. Komala, Y.-H. Chua and E. Iglesia, J. Catal., 2002, 205, 44–57 CrossRef CAS.
- B. L. Foley and A. Bhan, ACS Catal., 2020, 10, 10436–10448 CrossRef CAS.
- K. M. Keskinen, T. T. Pakkanen, P. Raulo, M. Ruotsalainen, P. Sarv and M. Tutta, in Studies in Surface Science and Catalysis, ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Hölderich, Elsevier, 1994, vol. 84, pp. 875–882 Search PubMed.
- P. J. Kunkeler, R. S. Downing and H. van Bekkum, Studies in Surface Science and Catalysis, Elsevier, 2001, vol. 137, pp. 987–1001 Search PubMed.
- H. P. Röger, K. P. Möller, W. Böhringer and C. T. O’Connor, Studies in Surface Science and Catalysis, Elsevier, 2000, vol. 130, pp. 2909–2914 Search PubMed.
- C. Li, H. J. Cho, Z. Wang, J. Gou, Y. Ren, H. Xi and W. Fan, ChemCatChem, 2016, 8, 2406–2414 CrossRef CAS.
- A. Korde, B. Min, Q. Almas, Y. Chiang, S. Nair and C. W. Jones, ChemCatChem, 2019, 11, 4548–4557 CrossRef CAS.
- L. Emdadi, S. C. Oh, Y. Wu, S. N. Oliaee, Y. Diao, G. Zhu and D. Liu, J. Catal., 2016, 335, 165–174 CrossRef CAS.
- H. I. Adawi, F. O. Odigie and M. L. Sarazen, Mol. Syst. Des. Eng., 2021, 6, 903–917 RSC.
- D. Xu, O. Abdelrahman, S. H. Ahn, Y. Guefrachi, A. Kuznetsov, L. Ren, S. Hwang, M. Khaleel, S. Al Hassan, D. Liu, S. B. Hong, P. Dauenhauer and M. Tsapatsis, AIChE J., 2019, 65, 1067–1075 CrossRef CAS.
- K. Hashimoto, T. Masuda and Y. Hariguchi, Nippon Kagaku Kaishi, 1989, 3, 575–582 CrossRef.
- D. Fraenkel, J. Catal., 1989, 118, 10–21 CrossRef CAS.
- A. W. Burton and S. I. Zones, in Studies in Surface Science and Catalysis, ed. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, 2007, vol. 168, pp. 137–179 Search PubMed.
- T. Ikuno, W. Chaikittisilp, Z. Liu, T. Iida, Y. Yanaba, T. Yoshikawa, S. Kohara, T. Wakihara and T. Okubo, J. Am. Chem. Soc., 2015, 137, 14533–14544 CrossRef CAS PubMed.
- J. E. Schmidt, D. Fu, M. W. Deem and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 16044–16048 CrossRef CAS PubMed.
- S. Li, R. Gounder, A. Debellis, I. B. Müller, S. Prasad, A. Moini and W. F. Schneider, J. Phys. Chem. C, 2019, 123, 17454–17458 CrossRef CAS.
- M. Kumar, Z. J. Berkson, R. J. Clark, Y. Shen, N. A. Prisco, Q. Zheng, Z. Zeng, H. Zheng, L. B. McCusker, J. C. Palmer, B. F. Chmelka and J. D. Rimer, J. Am. Chem. Soc., 2019, 141, 20155–20165 CrossRef CAS PubMed.
- Q. Ke, I. Khalil, B. Smeyers, Z. Li, R. de Oliveira-Silva, B. Sels, D. Sakellariou and M. Dusselier, Angew. Chem., Int. Ed., 2021, 60, 24189–24197 CrossRef CAS PubMed.
- S. I. Zones, K. Jayanthi, J. Pascual, D. Xie and A. Navrotsky, Chem. Mater., 2021, 33, 2126–2138 CrossRef CAS.
- D. Schwalbe-Koda, S. Kwon, C. Paris, E. Bello-Jurado, Z. Jensen, E. Olivetti, T. Willhammar, A. Corma, Y. Román-Leshkov, M. Moliner and R. Gómez-Bombarelli, Science, 2021, 374, 308–315 CrossRef CAS PubMed.
- C. Waitt, X. Gao, R. Gounder, A. Debellis, S. Prasad, A. Moini and W. F. Schneider, J. Phys. Chem. C, 2023, 127(46), 22740–22751 CrossRef CAS.
- F. Daeyaert and M. Deem, AI-Guided Design and Property Prediction for Zeolites and Nanoporous Materials, John Wiley & Sons, Ltd, 2023, pp. 33–59 Search PubMed.
- C. M. Lew, D. Xie, J. E. Schmidt, S. Elomari, T. M. Davis and S. I. Zones, AI-Guided Design and Property Prediction for Zeolites and Nanoporous Materials, John Wiley & Sons, Ltd, 2023, pp. 1–32 Search PubMed.
- J. Shin, N. H. Ahn, M. A. Camblor, S. J. Cho and S. B. Hong, Angew. Chem., Int. Ed., 2014, 53, 8949–8952 CrossRef CAS PubMed.
- J. Shin, N. H. Ahn, M. A. Camblor, C. M. Zicovich-Wilson and S. B. Hong, Chem. Mater., 2014, 26, 3361–3363 CrossRef CAS.
- J. Devos, S. Robijns, C. Van Goethem, I. Khalil and M. Dusselier, Chem. Mater., 2021, 33, 2516–2531 CrossRef CAS.
- S. Lee, C. T. Nimlos, E. R. Kipp, Y. Wang, X. Gao, W. F. Schneider, M. Lusardi, V. Vattipalli, S. Prasad, A. Moini and R. Gounder, Cryst. Growth Des., 2022, 22(10), 6275–6295 CrossRef CAS.
- H. I. Adawi, Y. Zheng and M. L. Sarazen, Cryst. Growth Des., 2023, 23, 2675–2688 CrossRef CAS.
- J. A. van Bokhoven, D. C. Koningsberger, P. Kunkeler, H. van Bekkum and A. P. M. Kentgens, J. Am. Chem. Soc., 2000, 122, 12842–12847 CrossRef CAS.
- T. Jarrin, T. de Bruin and C. Chizallet, ACS Catal., 2024, 1639–1652 CrossRef CAS.
- C. Lei, A. Erlebach, F. Brivio, L. Grajciar, Z. Tošner, C. J. Heard and P. Nachtigall, Chem. Sci., 2023, 14, 9101–9113 RSC.
- J. Martinez-Ortigosa, J. Simancas, J. A. Vidal-Moya, P. Gaveau, F. Rey, B. Alonso and T. Blasco, J. Phys. Chem. C, 2019, 123, 22324–22334 CrossRef CAS.
- H. Xiong, Z. Liu, X. Chen, H. Wang, W. Qian, C. Zhang, A. Zheng and F. Wei, Science, 2022, 376, 491–496 CrossRef CAS PubMed.
- B. Shen, H. Wang, H. Xiong, X. Chen, E. G. T. Bosch, I. Lazić, W. Qian and F. Wei, Nature, 2022, 1–5 CAS.
- B. Shen, X. Chen, H. Wang, H. Xiong, E. G. T. Bosch, I. Lazić, D. Cai, W. Qian, S. Jin, X. Liu, Y. Han and F. Wei, Nature, 2021, 592, 541–544 CrossRef CAS PubMed.
- H. Xiong, H. Wang, X. Chen and F. Wei, ACS Catal., 2023, 13, 12213–12226 CrossRef CAS.
- A. Hwang, T. T. Le, Z. Shi, H. Dai, J. D. Rimer and A. Bhan, J. Catal., 2019, 369, 122–132 CrossRef CAS.
- B. L. Foley, B. A. Johnson and A. Bhan, ACS Catal., 2021, 3628–3637 CrossRef CAS.
- N. K. Razdan, A. Kumar and A. Bhan, J. Catal., 2019, 372, 370–381 CrossRef CAS.
- Y. G. Hur, P. M. Kester, C. T. Nimlos, Y. Cho, J. T. Miller and R. Gounder, Ind. Eng. Chem. Res., 2019, 58, 11849–11860 CrossRef CAS.
- R. Kolvenbach, N. Al-Yassir, S. S. Al-Khattaf, O. C. Gobin, J. H. Ahn, A. Jentys and J. A. Lercher, Catal. Today, 2011, 168, 147–157 CrossRef CAS.
- T. Masuda, Catal. Surv. Asia, 2003, 7, 133–144 CrossRef CAS.
- R. Davis, AIChE J., 2018, 64, 3778–3785 CrossRef CAS.
- A. Bhan and W. N. Delgass, J. Catal., 2022, 405, 419–429 CrossRef CAS.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS.
- R. J. Quann, L. A. Green, S. A. Tabak and F. J. Krambeck, Ind. Eng. Chem. Res., 1988, 27, 565–570 CrossRef CAS.
- S. Ilias and A. Bhan, ACS Catal., 2013, 3, 18–31 CrossRef CAS.
- M. Moliner, J. M. Serra, A. Corma, E. Argente, S. Valero and V. Botti, Microporous Mesoporous Mater., 2005, 78, 73–81 CrossRef CAS.
- A. Corma, M. J. Díaz-Cabañas, J. L. Jordá, C. Martínez and M. Moliner, Nature, 2006, 443, 842–845 CrossRef CAS PubMed.
- A. Corma, M. Moliner, J. M. Serra, P. Serna, M. J. Díaz-Cabañas and L. A. Baumes, Chem. Mater., 2006, 18, 3287–3296 CrossRef CAS.
- R. Pophale, P. A. Cheeseman and M. W. Deem, Phys. Chem. Chem. Phys., 2011, 13, 12407–12412 RSC.
- K. Muraoka, Y. Sada, D. Miyazaki, W. Chaikittisilp and T. Okubo, Nat. Commun., 2019, 10, 4459 CrossRef PubMed.
- E. Pan, S. Kwon, Z. Jensen, M. Xie, R. Gómez-Bombarelli, M. Moliner, Y. Román-Leshkov and E. Olivetti, ACS Cent. Sci., 2024, 10(3), 729–743 CrossRef CAS PubMed.
- Z. Jensen, E. Kim, S. Kwon, T. Z. H. Gani, Y. Román-Leshkov, M. Moliner, A. Corma and E. Olivetti, ACS Cent. Sci., 2019, 5(5), 892–899 CrossRef CAS PubMed.
- Z. Jensen, S. Kwon, D. Schwalbe-Koda, C. Paris, R. Gómez-Bombarelli, Y. Román-Leshkov, A. Corma, M. Moliner and E. A. Olivetti, ACS Cent. Sci., 2021, 7(5), 858–867 CrossRef CAS PubMed.
- K. Muraoka, W. Chaikittisilp and T. Okubo, Chem. Sci., 2020, 11, 8214–8223 RSC.
- M. Moliner, Y. Román-Leshkov and A. Corma, Acc. Chem. Res., 2019, 52, 2971–2980 CrossRef CAS PubMed.
- J. Sauer, J. Catal., 2024, 433, 115482 CrossRef CAS.
- J. O. Hirschfelder, C. F. Curtiss and R. B. Bird, The Molecular Theory of Gases and Liquids, Wiley-Interscience, Hoboken, NJ, Revised edn, 1964 Search PubMed.
- D. W. Breck, Zeolite Molecular Sieves: Structure, Chemistry, and Use, Wiley, 1973 Search PubMed.
- C. E. Webster, R. S. Drago and M. C. Zerner, J. Am. Chem. Soc., 1998, 120, 5509–5516 CrossRef CAS.
- V. R. Choudhary, V. S. Nayak and T. V. Choudhary, Ind. Eng. Chem. Res., 1997, 36, 1812–1818 CrossRef CAS.
- M. L. Connolly, Science, 1983, 221, 709–713 CrossRef CAS PubMed.
- S. Herrmann and E. Iglesia, J. Catal., 2017, 346, 134–153 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |