
Photoinduced fluoroalkylation-peroxidation of alkenes enabled by ligand-to-iron charge transfer mediated decarboxylation†
Qiuwei
Huang
,
Chenhao
Lou
,
Leiyang
Lv
 * and
Zhiping
Li
*
* and
Zhiping
Li
*
Key Laboratory of Advanced Light Conversion Materials and Biophotonics, School of Chemistry and Life Resources, Renmin University of China, Beijing 100872, China. E-mail: lvleiyang2020@ruc.edu.cn; zhipingli@ruc.edu.cn
First published on 30th September 2024
Abstract
We report here a photoinduced iron-catalyzed fluoroalkylation-peroxidation of activated and/or unactivated alkenes with fluoroalkyl carboxylic acids and hydroperoxide. The ligand-to-iron charge transfer strategy effectively overcomes the high redox potential of the fluoroalkyl carboxylic acids, facilitating the difunctionalization reaction to occur smoothly under mild reaction conditions. The late-stage functionalization of drug and natural product derivatives was also demonstrated.
The incorporation of fluoroalkyl motifs into organic frameworks has emerged as a powerful strategy in the development of pharmaceuticals and agrochemicals. This approach can substantially enhance the therapeutic profiles of the parent compounds by improving their bioavailability, lipophilicity and metabolic stability.1 Among the various fluoroalkyl groups, the introduction of the trifluoromethyl (CF3) group, which functions as a valuable bioisostere for the methyl group, into aliphatic chains has gained increasing interest in recent years.2 In this regard, the radical fluoroalkylation of alkenes offers a straightforward approach to accessing a variety of trifluoromethylated products in a single step.3 To facilitate this transformation, several reagents have been developed, including Togni's reagent, Umemoto's reagent, Langlois’ reagent and Ruppert–Prakash reagent, all of which enable the efficient installation of the CF3 group. Nevertheless, the high cost of these reagents raises concerns regarding their accessibility and application.
Decarboxylative functionalization of carboxylic acids represents research in the field of synthetic chemistry.4 Among the various candidates, trifluoroacetic acid (TFA) is of particular interest due to its stability and low cost, which makes it an attractive option to achieve trifluoromethylation. Theoretically, TFA can undergo single-electron transfer (SET) oxidation to generate a CF3 radical, accompanied by the release of CO2. However, carboxylates bearing strong electron-withdrawing fluoroalkyl substituents display an exceptionally high redox potential for CF3COO− [Ep/2 > 2.28 V vs. a saturated calomel electrode (SCE)].5 This elevated redox potential makes the seemingly straightforward decarboxylative pathway quite challenging. Consequently, the direct outer-sphere electron oxidation of TFA necessitates the use of harsh reaction conditions, such as elevated temperatures (>120 °C), and the use of strong oxidants such as XeF2.6
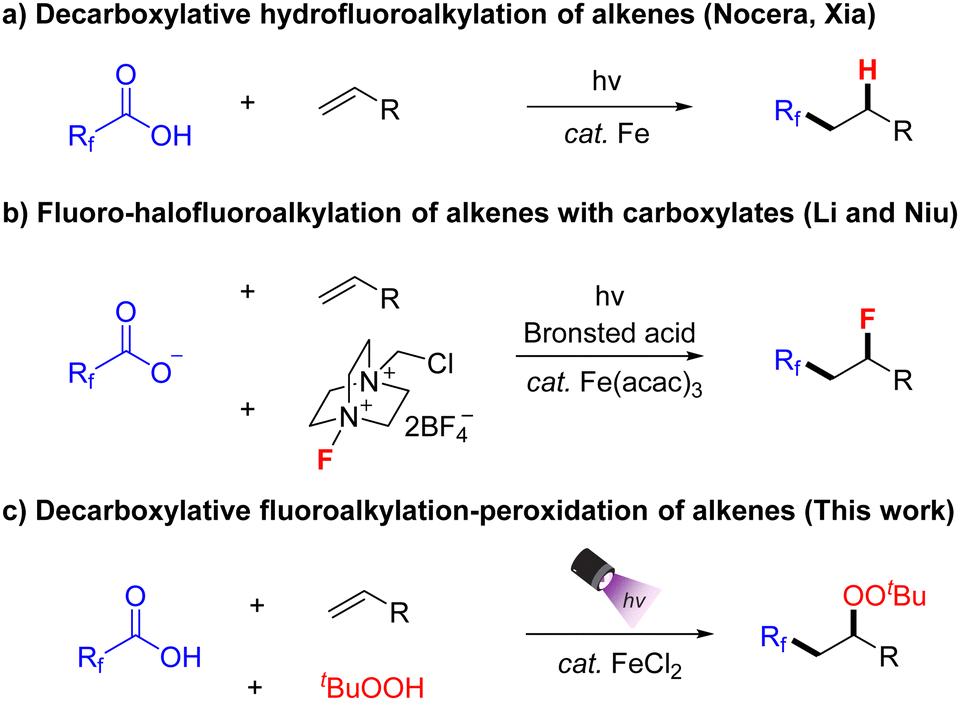
Photocatalysis has emerged as a powerful approach to facilitate challenging organic transformations under mild reaction conditions.7 Among the various photocatalytic strategies, ligand-to-metal charge transfer (LMCT) processes have been used to generate radical species from inactive substrates. As a representative inner-sphere single electron transfer mechanism, LMCT enables electron transfer to or from a directly coordinated substrate and a metal, irrespective of the redox potentials of the substrates and the photocatalyst.8 In 2023, West and coworkers reported a state-of-the-art iron-mediated photocatalytic hydrofluoroalkylation of alkenes with diverse fluoroalkyl carboxylic acids via LMCT and a thiol-mediated HAT process (Scheme 1a).9 Based on the Fe-LMCT strategy, Xia and coworkers also achieved a similar transformation shortly thereafter, demonstrating the feasibility of performing hydrofluoroalkylation on a gram-scale in conjunction with continuous flow synthesis.10 In 2024, Niu, Li and co-workers further developed this strategy by developing a Brønsted acid-mediated Fe-LMCT activation of inert fluoroalkylcarboxylates, which enabled the fluoro-polyfluoroalkylation of alkenes with Selectfluor as the fluorine source (Scheme 1b).11 Despite these achievements, the incorporation of a CF3 group and another functional group (e.g. difunctionalization) into alkenes using TFA is rather limited.
 | ||
| Scheme 1 Ligand-to-iron charge transfer enabled fluoroalkylation-functionalization of alkenes. | ||
Organic peroxides play a vital role in a number of fields, particularly in the large-scale production of polymers and in medicinal chemistry.12 In continuation of our research interests in iron catalysis13 as well as the synthesis and manipulation of organic peroxides,14 we herein report the photocatalytic fluoroalkylation-peroxidation of alkenes using an iron-LMCT strategy (Scheme 1c). This cooperative catalytic system enables the efficient implementation of the three-component reaction involving fluoroalkyl carboxylic acids, alkenes and tert-butyl hydroperoxide, affording a variety of fluoroalkylated organic peroxides in moderate to good yields under mild reaction conditions.
We initiated the investigation with the use of 4-phenylbutene 1a, TFA 2a and tert-butyl hydroperoxide 3 as the model substrates to test our hypothesis (Table 1). Encouragingly, the desired trifluoromethylation-peroxidation product 4a was obtained in 32% yield with Fe(acac)3 as the catalyst and a catalytic amount of Na2CO3 in MeCN (0.1 M) under 390![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm irradiation at room temperature for 24 h (entry 1). Various iron catalysts, including Fe(OTf)2, FeCl3, Fe(NO3)3·9H2O and Fe(OAc)2, FeCl2 were tested (entries 2–6). Among these, FeCl2 gives the highest yield of 4a at 79% (entry 6). Subsequently, we tested different solvents, including ethyl acetate (EtOAc), dichloromethane (DCM), 1,2-dichloroethane (DCE), methyl tert-butyl ether (MTBE), benzene and dimethylsulfoxide (DMSO) (entries 7–12), and found that MeCN remained the optimal solvent. The desired product 4a could also be synthesized in the presence of alternative bases, such as Li2CO3, K2CO3, Cs2CO3, N,N-diisopropylethylamine (DIPEA) and 1,4-diazabicyclo[2.2.2]octane (DABCO) (entries 13–17). However, the yields were inferior compared to those achieved with Na2CO3. The product 4a was obtained in 40% yield when tBuOOH (70% in H2O) was used instead (entry 18). Control experiments confirmed that 4a was not detected in the absence of a metal catalyst (entry 19) or light (entry 20).
nm irradiation at room temperature for 24 h (entry 1). Various iron catalysts, including Fe(OTf)2, FeCl3, Fe(NO3)3·9H2O and Fe(OAc)2, FeCl2 were tested (entries 2–6). Among these, FeCl2 gives the highest yield of 4a at 79% (entry 6). Subsequently, we tested different solvents, including ethyl acetate (EtOAc), dichloromethane (DCM), 1,2-dichloroethane (DCE), methyl tert-butyl ether (MTBE), benzene and dimethylsulfoxide (DMSO) (entries 7–12), and found that MeCN remained the optimal solvent. The desired product 4a could also be synthesized in the presence of alternative bases, such as Li2CO3, K2CO3, Cs2CO3, N,N-diisopropylethylamine (DIPEA) and 1,4-diazabicyclo[2.2.2]octane (DABCO) (entries 13–17). However, the yields were inferior compared to those achieved with Na2CO3. The product 4a was obtained in 40% yield when tBuOOH (70% in H2O) was used instead (entry 18). Control experiments confirmed that 4a was not detected in the absence of a metal catalyst (entry 19) or light (entry 20).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Yield of 4a |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), tBuOOH 3 (0.8 mmol, 5–6 M in decane), catalyst (10.0 mol%), base (10.0 mol%), solvent (2.0 mL), 390 nm (10 W), r.t., 24 h in a Schlenk tube under N2. b NMR yields are based on 1a and determined by 1H NMR using mesitylene as an internal standard. c In the dark. d t BuOOH (70% in H2O). | ||||
| 1 | Fe(acac)3 | Na2CO3 | MeCN | 32 |
| 2 | Fe(OTf)2 | Na2CO3 | MeCN | 31 |
| 3 | FeCl3 | Na2CO3 | MeCN | 58 |
| 4 | Fe(NO3)3·9H2O | Na2CO3 | MeCN | 65 |
| 5 | Fe(OAc)2 | Na2CO3 | MeCN | 71 |
| 6 | FeCl 2 | Na 2 CO 3 | MeCN | 79 |
| 7 | FeCl2 | Na2CO3 | EtOAc | 36 |
| 8 | FeCl2 | Na2CO3 | DCM | 0 |
| 9 | FeCl2 | Na2CO3 | DCE | 0 |
| 10 | FeCl2 | Na2CO3 | MTBE | 15 |
| 11 | FeCl2 | Na2CO3 | Benzene | 0 |
| 12 | FeCl2 | Na2CO3 | DMSO | 0 |
| 13 | FeCl2 | Li2CO3 | MeCN | 60 |
| 14 | FeCl2 | K2CO3 | MeCN | 45 |
| 15 | FeCl2 | Cs2CO3 | MeCN | 38 |
| 16 | FeCl2 | DIPEA | MeCN | 60 |
| 17 | FeCl2 | DABCO | MeCN | 58 |
| 18d | FeCl2 | Na2CO3 | MeCN | 40 |
| 19 | — | Na2CO3 | MeCN | 0 |
| 20c | FeCl2 | Na2CO3 | MeCN | 0 |

With the optimal reaction conditions established, we set out to investigate the generality of this protocol. As shown in Scheme 2, a variety of fluorinated peroxides were synthesized from unactivated alkenes in moderate to good yields. The functional groups, including ester 1b, ketone 1c, alcohol 1d, bromide 1e, terminal alkyne 1f, N-phthalimide 1g and phosphate 1h were well tolerated, affording the corresponding products 4b–4h in 41–88% yields. The reaction also proceeded with alkenes containing heterocycles such as thiophene 1i, furan 1j and pyridine 1k, affording the desired products 4i–4k. Additionally, cyclobutene carboxylic acid derivative 1l and 1-dodecene 1m proceeded smoothly, producing the desired products 4l and 4m in 88% and 68% yields, respectively. Styrene derivatives 1n–1p, featuring either electron-donating or electron-withdrawing groups, were successfully converted into the corresponding fluorinated peroxides 4n–4p in 47–74% yields. Other types of alkenes, including enyne 1r, acrylate 1q and allyl ether 1s, also underwent this iron-mediated photocatalytic trifluoromethylation-peroxidation reaction, although the yields were relatively low (25–33%), probably due to polymerization or other side reactions under 390 nm irradiation. Moreover, 1,1-disubstituted alkenes were suitable substrates as well, affording the corresponding difluorinated peroxides 4t–4x in 34–77% yields. We also tested a series of internal alkenes under the optimal conditions. Cyclic alkenes, including cyclohexene 1y, cyclooctene 1z, norbornene 1aa and indene 1ab, provided the corresponding products 4y–4ab in 19–66% yields. Besides, the peroxides 4ac–4ae were obtained in 25–46% yields when 1,2-disubstituted or trisubstituted acyclic alkenes were tested accordingly.
 | ||
| Scheme 2 Scope of substrates. aAlkene 1 (0.2 mmol), RfCOOH 2 (0.6 mmol), tBuOOH 3 (0.8 mmol, 5.5 M in decane), FeCl2 (10% mol) and Na2CO3 (0.02 mmol) in MeCN (2.0 mL), 390 nm Kessil LED at r.t. for 24 h under N2. d.r. = diastereoselectivity ratio. | ||
In light of our findings regarding the use of trifluoroacetic acid for the trifluoromethylation-peroxidation of alkenes, we proceeded to investigate the potential of other fluoroalkylated carboxylic acids as reaction partners. For example, difluoroacetic acid 2b, 2,2-difluoropropanoic acid 2c, perfluorooctanoic acid 2d, perfluorooctanoic acid 2d and phenyl difluoroacetic acid 2e were identified as suitable candidates, affording the desired products 4af–4al in 29–79% yields.
To demonstrate the practical applicability of this protocol, we investigated its potential for late-stage functionalization of drug and natural product derivatives (Scheme 3). The unactivated alkenes derived from various precursors, including sugar, non-steroidal anti-inflammatory and analgesic drugs such as celecoxib, sulbactam, gemfibrozil, as well as steroidal compounds like dehydrocholic acid, were reacted efficiently, providing the corresponding fluorinated peroxides 6a–6e in 51–79% yields. Moreover, (L)-menthol and the sterically congested (−)-borneol derivatives could also be synthesized efficiently (6f and 6g).
 | ||
| Scheme 3 Modification of drug or natural product derivatives. | ||
The photoinduced fluoroalkylation-peroxidation reaction was completely inhibited in the presence of radical scavengers, such as TEMPO (2,2,6,6-tetramethylpiperidin-1-oxyl). Moreover, the radical clock experiment gave the ring-opening fluorinated peroxide in 48% yield (see the ESI† for details). These findings suggest that a radical pathway was involved in this process.
Based on the above results and literature reports,9–11,14 a plausible reaction mechanism for the fluoroalkylation-peroxidation of alkenes is presented in Scheme 4. Initially, Fe(II) is oxidized by tBuOOH to give Fe(III) and tBuO˙. The photoactive Fe(III)-carboxylate complex A is formed in situ via the coordination of fluoroalkyl carboxylic acid 2 with Fe(III). Upon photoexcitation, a LMCT event occurs to generate Fe(II) and an acyloxy radical B, which subsequently undergoes decarboxylation to release the fluoroalkyl radical C. The electrophilic radical C is then trapped by an alkene, leading to the formation of the nucleophilic radical intermediate D. Concurrently, tBuO˙ abstracts a hydrogen atom from tBuOOH, producing the tBuOO˙ E. Finally, a polarity-matched radical coupling between radicals D and E results in the formation of the fluorinated peroxide product 4.
 | ||
| Scheme 4 A plausible reaction mechanism. | ||
In conclusion, we have developed a photocatalytic fluoroalkylation-peroxidation of alkenes utilizing an iron-LMCT strategy. The cooperative catalytic system is effective in facilitating a three-component reaction involving fluoroalkyl carboxylic acids, alkenes and tert-butyl hydroperoxide. A diverse array of fluoroalkylated organic peroxides were synthesized in moderate to good yields under mild reaction conditions. Furthermore, the late-stage functionalization of drug and natural product derivatives was successfully accomplished.
We gratefully acknowledge the National Natural Science Foundation of China (22071266, 22201300), the Fundamental Research Funds for the Central Universities, and the Research Funds of Renmin University of China (24XNKJ27).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews, see: (a) W. R. Dolbier, Chem. Rev., 1996, 96, 1557–1584 CrossRef PubMed; (b) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1–PR43 CrossRef PubMed; (c) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC; (d) W. Wu, Y. You and Z. Weng, Chin. Chem. Lett., 2022, 33, 4517–4530 CrossRef.
- For selected reviews, see: (a) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef PubMed; (b) C. Alonso, E. Martinez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef PubMed; (c) H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC; (d) W. C. Fu, P. M. MacQueen and T. F. Jamison, Chem. Soc. Rev., 2021, 50, 7378–7394 RSC.
- (a) H. Xiao, H. Shen, L. Zhu and C. Li, J. Am. Chem. Soc., 2019, 141, 11440–11445 CrossRef CAS PubMed; (b) X. Dong, W. Jiang, D. Hua, X. Wang, L. Xu and X. Wu, Chem. Sci., 2021, 12, 11762–11768 RSC; (c) N. E. Intermaggio, A. Millet, D. L. Davis and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 11961–11968 CrossRef CAS; (d) S. Fernandez-Garcia, V. O. Chantzakou and F. Julia-Hernandez, Angew. Chem., Int. Ed., 2024, 63, e202311984 CrossRef CAS PubMed; (e) B. Wang, Y. Xu, J. Wang, M. Yang, G.-J. Deng and W. Shao, Chin. Chem. Lett., 2024, 35, 109502 CrossRef CAS; (f) X.-Y. Cheng, Y.-F. Zhang, J.-H. Wang, Q.-S. Gu, Z.-L. Li and X.-Y. Liu, J. Chem. Am. Soc., 2022, 144, 18081–18089 CrossRef CAS; (g) Z. Li, L. Jiao, Y. Sun, Z. He, Z. Wei and W.-W. Liao, Angew. Chem., Int. Ed., 2020, 59, 7266–7270 CrossRef CAS.
- (a) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D. H. Bao, F. L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS; (b) D. M. Kitcatt, S. Nicolle and A. L. Lee, Chem. Soc. Rev., 2022, 51, 1415–1453 RSC; (c) P. Xiao, X. Pannecoucke, J. P. Bouillon and S. Couve-Bonnaire, Chem. Soc. Rev., 2021, 50, 6094–6151 RSC.
- (a) C. Depecker, H. Marzouk, S. Trevin and J. Devynck, New J. Chem., 1999, 23, 739–742 RSC; (b) D. Nicewicz, H. Roth and N. Romero, Synlett, 2015, 714–723 CrossRef.
- (a) M. Chen and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 11628–11631 CrossRef CAS; (b) B. Yang, D. Yu, X.-H. Xu and F.-L. Qing, ACS Catal., 2018, 8, 2839–2843 CrossRef CAS; (c) J. Qi, J. Xu, H. T. Ang, B. Wang, N. K. Gupta, S. R. Dubbaka, P. O‘Neill, X. Mao, Y. Lum and J. Wu, J. Am. Chem. Soc., 2023, 145, 24965–24971 CAS.
- (a) D. Staveness, I. Bosque and C. R. Stephenson, Acc. Chem. Res., 2016, 49, 2295–2306 CrossRef CAS; (b) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (c) P. Z. Wang, W. J. Xiao and J. R. Chen, Nat. Rev. Chem., 2023, 7, 35–50 CrossRef PubMed.
- (a) Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS; (b) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef PubMed; (c) A. M. May and J. L. Dempsey, Chem. Sci., 2024, 15, 6661–6678 RSC.
- K. J. Bian, Y. C. Lu, D. Nemoto, Jr., S. C. Kao, X. Chen and J. G. West, Nat. Chem., 2023, 15, 1683–1692 CrossRef.
- (a) X.-K. Qi, L.-J. Yao, M.-J. Zheng, L. Zhao, C. Yang, L. Guo and W. Xia, ACS Catal., 2024, 14, 1300–1310 CrossRef; (b) Z. Song, L. Guo, C. Yang and W. Xia, Org. Chem. Front., 2024, 11, 4436–4441 RSC; (c) M. Luo, S. Zhu, J. Yin, C. Yang, L. Guo and W. Xia, Org. Chem. Front., 2024, 11, 4748–4756 RSC.
- X. Jiang, Y. Lan, Y. Hao, K. Jiang, J. He, J. Zhu, S. Jia, J. Song, S. J. Li and L. Niu, Nat. Commun., 2024, 15, 6115 CrossRef CAS PubMed.
- (a) J. K. Cheng and T. P. Loh, J. Am. Chem. Soc., 2015, 137, 42–45 CrossRef CAS PubMed; (b) B. Schweitzer-Chaput, J. Demaerel, H. Engler and M. Klussmann, Angew. Chem., Int. Ed., 2014, 53, 8737–8740 CrossRef CAS PubMed; (c) I. A. Yaremenko, V. A. Vil’, D. V. Demchuk and A. O. Terent’ev, Beilstein J. Org. Chem., 2016, 12, 1647–1748 CrossRef CAS PubMed; (d) N. Zhu, H. Yao, X. Zhang and H. Bao, Chem. Soc. Rev., 2024, 53, 2326–2349 RSC.
- (a) F. Jia and Z. Li, Org. Chem. Front., 2014, 1, 194–214 RSC; (b) L. Lv and Z. Li, Top. Curr. Chem., 2016, 374, 38 CrossRef PubMed.
- (a) W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756–10759 CrossRef CAS; (b) L. Lv, B. Shen and Z. Li, Angew. Chem., Int. Ed., 2014, 53, 4164–4167 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR and 13C NMR spectra for new compounds. See DOI: https://doi.org/10.1039/d4cc04650a |
| This journal is © The Royal Society of Chemistry 2024 |