
Design and synthesis of piperine-based photoaffinity probes for revealing potential targets of piperine in neurological disease†
Li
Shen‡
ab,
Yue
Yang‡
ab,
Lijun
Lu
 c,
Jili
Huang
ab,
Wen
He
ab,
Chunfang
Zhao
ab,
Feng
Guo
ab,
Chunbo
Zhang
*ab,
Haijun
Zhong
*ab and
Fan
Liao
*ab
c,
Jili
Huang
ab,
Wen
He
ab,
Chunfang
Zhao
ab,
Feng
Guo
ab,
Chunbo
Zhang
*ab,
Haijun
Zhong
*ab and
Fan
Liao
*ab
aDepartment of Pharmaceutics, College of Pharmacy, Nanchang University, Nanchang 330006, Jiangxi, China. E-mail: liaofan@ncu.edu.cn; zhonghj@ncu.edu.cn; cbzhang@ncu.edu.cn
bThe MOE Basic Research and Innovation Center for the Targeted Therapeutics of Solid Tumors, Jiangxi Province Key Laboratory of Drug Target Discovery and Validation, Jiangxi Medical College, Nanchang University, Nanchang 330031, China
cDepartment of Molecular Chemistry and Materials Science, Weizmann Institute of Science, Rehovot 7610001, Israel
First published on 29th November 2024
Abstract
Piperine (PIP) has attracted extensive attention due to its diverse biological activities. In this study, we developed two photoaffinity probes PIP-1 and PIP-2, which are biologically safe and retain PIP's bioactivity, to investigate its protein targets in vivo. Using in situ labeling and cell imaging, we were able to effectively detect and visualize the drug targets of PIP with our probes. Additionally, a series of protein targets of PIP were fished using PIP-2 through proteome profiling, with further validation suggesting that TGFβ1 might be a potential target involved in PIP's effects on neurological diseases. These findings demonstrate that PIP-2 is a valuable tool for identifying the targets of PIP.
Piperine (PIP) is an alkaloid extracted from Piperaceae plants, and it has been shown to exhibit a broad spectrum of biological activities.1–3 PIP demonstrates therapeutic efficacy in neurological diseases. For example, when combined with quercetin, PIP could enhance antioxidant, anti-inflammatory, and neuroprotective effects on rotenone-induced Parkinson's disease model rats.4 PIP could attenuate cognitive impairment in an experimental mouse model of sporadic Alzheimer's disease.5,6 Mechanistic investigation revealed that the antidepressant effects of PIP may be attributed to the activation of 5-HT1A and 5-HT1B receptors, resulting in increased 5-HT levels in the hippocampus and prefrontal cortex.7,8 Recent studies have shown that PIP can significantly enhance the bioavailability and absorption of carbamazepine, while also shortening its half-life in patients with epilepsy.9,10 These findings highlight the promising potential of PIP in the treatment of neurological disorders. However, the specific targets and neuropharmacological mechanism of PIP remain unclear due to its multi-target and pleiotropic effects, which limits its clinical application in neurotherapy. Therefore, identifying the key targets of PIP in neurological diseases is essential for advancing its therapeutic potential.
In recent years, the affinity-based protein profiling (AfBPP) technique has emerged as a powerful tool for identifying the protein targets of various bioactive small molecules.11,12 Photoaffinity probes can be covalently attached to target proteins in situ through functional groups, allowing for the monitoring of ligand-protein interactions in situ and the environment of these binding proteins for subsequent identification.13,14 Importantly, designing the chemical probe for target fishing is crucial for AfBPP. The drug must be modified with a functional group that allows it to capture its targets. A well-designed probe should preserve the biological activity of the drug,15 therefore, it is essential to minimize modifications to the drug molecule and carefully select the optimal site for modification.11,16 So far, in situ target identification using AfBPP has been successfully applied to numerous natural products, such as bile acid, tetrahydrocannabinol, and 7-oxocallitrisic acid.17–22 Hence, applying the AfBPP technique to investigate the potential targets of PIP holds great promise.
In this work, we aimed to develop effective photoaffinity probes to explore the PIP's targets. PIP is composed of three main structural components: a methylenedioxyphenyl (MDP) ring, a butadiene chain, and an amide with a piperidine ring (Fig. 1).
 | ||
| Fig. 1 Chemical structure of PIP and structures of two PIP photoaffinity probes. (Left) The structure of PIP. (Right) Two PIP photoaffinity probes, PIP-1 and PIP-2. | ||
Previous studies have shown that the double bond and tertiary amide bond in PIP are essential for its biological activity. Interestingly, replacing the amide bond with an easter bond has also been found to maintain the neuroactivity of PIP.23–27 Therefore, we selected the piperidine ring as the modification site to retain the key active groups in PIP. Based on this, we designed and synthesized two photoaffinity probes: PIP-1 with ester group and PIP-2 with maintaining tertiary amide group (Fig. 1). The “minimalist” bioorthogonal handles with photo-crosslinkers,28,29 which are widely used to efficiently fish out the actual targets of a drug and distinguish between covalent and non-covalent binding interactions, were incorporated into the PIP core structure. In PIP-1/2, the diazirine group, as the photoaffinity group, can form covalent bonds with adjacent proteins under ultraviolet light, thereby enhancing the stability of the probe-protein complex. On the other hand, the alkynyl group is used as the bioorthogonal handle, for subsequent binding to a reporter group, enabling protein labeling and target-capture analysis. PIP-1/2 was synthesized following the steps shown in the Scheme 1. The “minimalist” bioorthogonal handles with photo-crosslinkers compound 5 and compound 7 (Scheme 1A), were synthsized firstly and then reacted with piperic acid to produce PIP-1 and PIP-2 (Scheme 1B, see details in ESI†).
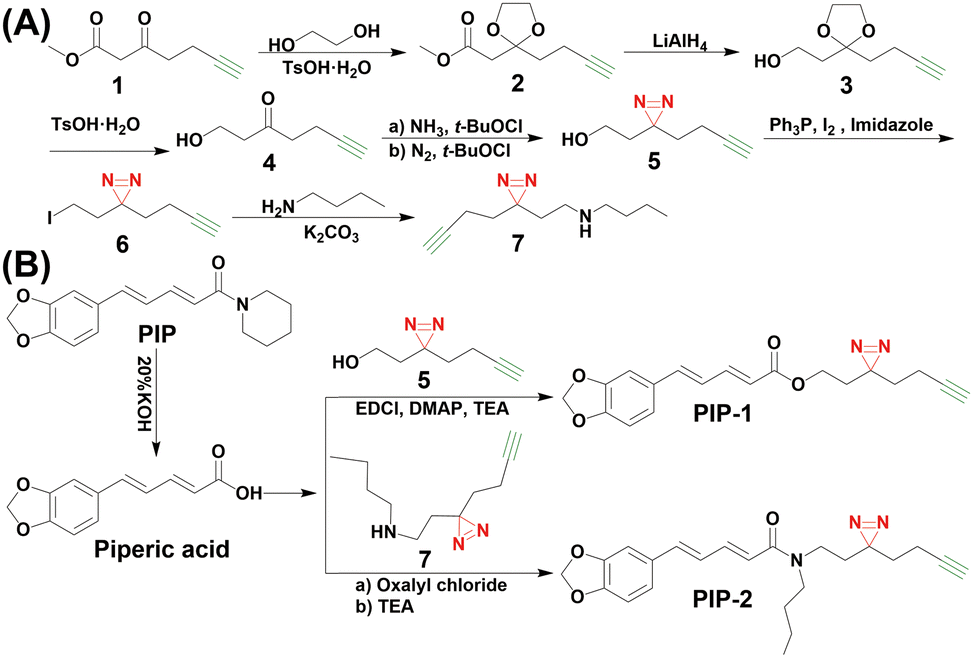 | ||
| Scheme 1 Synthetic route to PIP probes. | ||
Astrocytes were chosen as the cell type to study the protein targets due to their high abundance in the central nervous system (CNS) and their critical role in various neurological disorders.30,31 After synthesizing the probes, we assessed the biological safety of PIP-1/2 by evaluating their impact on astrocytes (CTX-TNA2 cell) viability using a CCK8 assay. As shown in Fig. S1 (ESI†), the concentration range of 0.1–100 μM had no significant influence on the activity of either PIP or PIP-1/2, indicating that PIP and PIP-1/2 have a favorable safety profile in CTX-TNA2 cells. As we known, the antiepileptic effect of PIP has been demonstrated in both preclinical and clinical studies.10,32 Accordingly, we assessed the biological activity of PIP-1/2 by evaluating their anticonvulsant efficacy. We used the pentylenetetrazole (PTZ)-induced zebrafish epilepsy model, one of the most widely used animal models for seizure testing,33 to evaluate the anticonvulsant effects of PIP-1/PIP-2. As shown in Fig. 2A, compared to the PTZ group, we observed a significant reduction in total travel distance when different concentrations of PIP-1/2 and PIP were introduced to the PTZ-containing culture medium. These results suggest that PIP-1/2 retain the antiepileptic activity of PIP, with PIP-2 exhibiting superior efficacy than PIP-1. Overall, these findings demonstrate that PIP-1/2 possess good biosafety and maintain bioactivity.
 | ||
| Fig. 2 (A) Evaluation of antiepileptic activity of PIP-1/2 probes, data presented by the average movement of different zebrafish groups within 20 mins relative to the PTZ group ± S.D. (n = 8, *p < 0.05, **p < 0.005, ****p < 0.0001, compared with PTZ group). (B) Gel-based profiling of PIP-1/2 labeled proteomes of CTX-TNA2 cell lines in situ (living cells): fluorescence scanning (Rhodamine, upper) and Coomassie Blue staining (CBB, lower). (C) Live cell imaging of CTX-TNA2 cells with PIP-1/2. Blue: Hoechst nuclear stain; green: Rhodamine channel. Scale bar: 10 μm. (D) Comparison of single cell fluorescence intensity of Rhodamine-N3 green fluorescence channel between probe group and competing group by Image Pro Plus 6.0. N = 5 (number of cells), *p < 0.05, ****p < 0.0001. | ||
Next, in situ labeling experiments were performed in astrocytes (Scheme S2, ESI†). The labeling results, shown in Fig. 2B, demonstrate that PIP-1 and PIP-2 effectively label proteins with a molecular weight of approximately 35 KDa, with labeling dependent on the probe concentration. Additionally, we observed that probe-induced labeling could be inhibited by pretreating the proteome sample with excess PIP. This competition assay indicated that PIP competes with PIP-1/2 for protein-binding sites, inhibiting their interaction with intracellular proteins. This result suggests that the probe molecule share the same target as PIP in vivo. Moreover, the intensity of labeled bands for PIP-1/2 significantly decreased without ultraviolet irradiation, demonstrating that PIP primarily binds to its target protein through non-covalent interactions.
The mode of action and target distribution of PIP and PIP-1/2 in vivo were further investigated through cellular imaging analysis. As shown in Fig. 2C, the blue fluorescence in Hoechst group represents the nucleus stain, while the green fluorescence in rhodamine-N3 group is from PIP-1/PIP-2. The imaging results reveal that the fluorescence signal of the probe group is predominantly localized outside the nucleus. However, upon competition with PIP, the extranuclear fluorescence signal in both PIP-1/PIP and PIP-2/PIP groups was significantly reduced, with a more pronounced decrease observed in the PIP-2/PIP group. Semi-quantitative analysis of fluorescence intensity is presented in Fig. 2D. These results suggest that PIP-1/2 competes with PIP for binding to intracellular targets, indicating a shared target between PIP-1/2 and PIP. Additionally, we observed a superior fluorescent labeling effect for PIP-2 compared to PIP-1, highlighting the crucial role of the amide bond in preserving PIP's activity.
Based on the labeling results and cellular imaging analysis, we identified the protein targets of PIP-2 through pull-down enrichment followed by LC–MS/MS analysis (Scheme S3, ESI†). To minimize variations introduced by the stochastic nature of the LC-MS/MS data-dependent acquisition, only proteins identified in all three replicates were considered for further analysis (Table S1–S3, ESI†). Proteins labeled by DMSO and those detected upon addition of 10 × PIP were excluded, resulting in a set of high-confidence target proteins (Fig. 3A). Fig. 3B shows the key proteins identified as interacting with PIP-2 with a molecular weight of around 35 KDa. We then performed subcellular localization analysis of these PIP-interacting proteins (Fig. S2, ESI†). The results indicated that the majority of PIP-2 target proteins were localized in the cytoplasm (48%), followed by secretory proteins (13%) and plasma membrane proteins (2%). Notably, these targets were predominantly located outside the nucleus, which aligns with the fluorescence imaging observations. We also queried the Kyoto Encyclopedia of Genes and Genomes (KEGG) database to categorize the interacting proteins based on the functional pathways they are involved in (Fig. S3, ESI†). The clustering analysis revealed that the most significantly enriched pathways included tight junction, motor proteins, the IL-17 signaling pathway, the Hippo signaling pathway, among others.
 | ||
| Fig. 3 (A) Target overlap between PIP-2 (red), PIP-2/PIP (green), and control (purple) cells. (B) Proteomic analysis of primary proteins that interacted with PIP-2. | ||
Among the protein we obtained, transforming growth factor beta (TGF-β) has been identified as one of the key factors regulating astrocyte reactivity.34 Therefore, we selected TGFβ1 for western blot analysis to validate it as a PIP target. As shown in Fig. 4A and Fig. S4 (ESI†), endogenous TGFβ1 was specifically labeled by PIP-2, and the intensity of the protein bands significantly decreased in the presence of PIP. These findings confirm an in vivo interaction between PIP and TGFβ1, supporting TGFβ1 as a potential target for PIP.
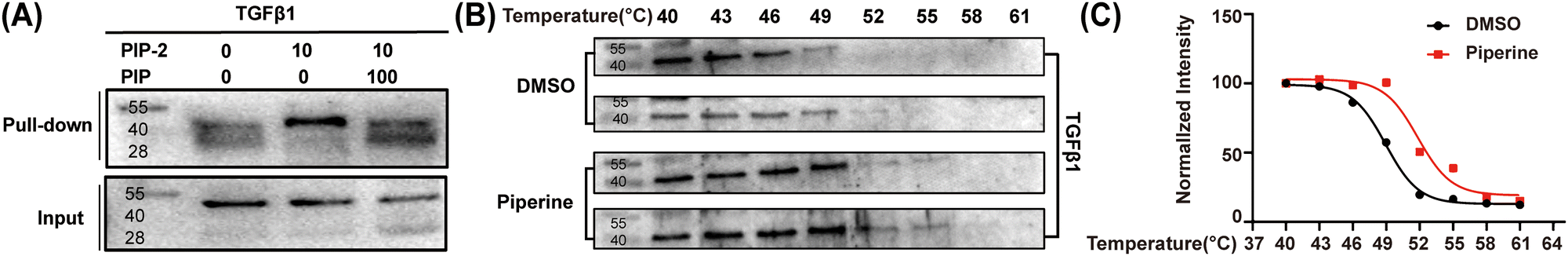 | ||
| Fig. 4 (A) Biotin-labeled target proteins identified using immunoblotting. (B) CETSA was used to evaluate the binding between PIP and TGFβ1 in thermodynamic levels (n = 2). (C) The melting curve of TGFβ1 with PIP. | ||
To further validate the binding of PIP to TGFβ1, we performed cellular thermal shift analysis (CETSA). Compared to DMSO control, PIP-treated cells exhibited a rightward shift in the melting curve (Fig. 4B, C, Fig. S5, Scheme S4, ESI†), indicating that PIP stabilized TGFβ1. To investigate the ligand-binding site, we simulated the binding of PIP to the crystal structure of TGFβ1 (PDB: 4KV535) using AutoDock Vina software.36 Our observation reveals that PIP forms hydrogen bonds with ARG-241, GLN-43, GLY-42, and ALA-40 (Fig. 5). Previous literatures have highlighted the potential of PIP in regulating TGFβ1 to exert anti-inflammatory and anti-fibrotic effects.37,38 Activation of the TGF-β/Smad signaling pathway has been shown to alleviate neuronal apoptosis and reduce symptoms of epilepsy.39 And the activation of TGF-β activated kinase 1 (TAK1) in microglia after experimental epilepsy contributes to epileptogenesis.40 Here, we have confirmed the direct binding of PIP to TGFβ1, suggesting that TGFβ1 may serve as a promising target for PIP in the context of neurological diseases treatment and drug discovery.
 | ||
| Fig. 5 Proposed binding mode of compound PIP to TGFβ1. | ||
In conclusion, we have designed and synthesized two photoaffinity probes, PIP-1 and PIP-2, specifically for investigating the targets of PIP. The results demonstrate that both probes exhibit excellent biosafety and bioactivity. Furthermore, the probes are effective in labeling proteins in nerve cells. The superior performance of PIP-2 as a probe was demonstrated by its increased target overlap with PIP, suggesting that the tertiary amide moiety is essential for PIP's biological activity. Results also indicate that PIP primarily interacts with its target proteins through non-covalent binding. Additionally, the targets of PIP are predominantly localized outside the nucleus. Using probe PIP-2, a series of protein targets were identified through proteome profiling, with TGFβ1 emerging as a potential target for studying the effects of PIP on neurological disease. Overall, this study demonstrates the feasibility of using PIP-1/2 as photoaffinity probes to investigate the targets of PIP, providing a valuable tool for elucidating the pharmacologic mechanism of PIP. However, in the current form, the bioorthogonal reaction used here is catalyzed by metal catalyst, which is not suitable for in vivo imaging systems. Moreover, the probe was obtained by modifying the PIP structure. Therefore, the targets detected by PIP-1/PIP-2 cannot fully overlap with those detected by PIP. But we think that these challenges can be overcome by future developments in the bioorthogonal chemistry and a highly accurate nonlabeling chemical proteomics approach with high-throughput. Further research is needed to focus on exploring potential targets in the other nerve cells and establishing the targets network of PIP by network pharmacological analysis, so as to find the targets and their specific connection for neurological disease.
This work was supported by Cultivation Project of Nanchang University, the Nanchang University Pilot and Demonstration Project of Contract Responsibility System of the Special Financial Fund of Science and Technology of Jiangxi Province of China (Grant No. ZBG20230418038), as well as the technical assistance offered by the instrument platform of Nanchang University and the School of Pharmacy. We thank Dr Fangling Lu (Jiangxi University of Traditional Chinese Medicine), Guoxing Deng, Jinyin Yi for their assistance with the experiments.
Data availability
The data supporting this article have been included as part of the ESI.† Venn diagrams was generated at https://www.bioinformatics.psb.ugent.be/webtools/Venn/. The crystal structure of TGFβ1 (4KV5) from https://www.rcsb.org/.Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Zadorozhna, T. Tataranni and D. Mangieri, Mol. Biol. Rep., 2019, 46, 5617–5629 CrossRef CAS.
- S. Azam, J. Y. Park, I. S. Kim and D. K. Choi, Biomedicines, 2022, 10, 154 CrossRef CAS PubMed.
- A. H. Tantawy, K. A. Soliman and H. M. Abd El-Lateef, J. Clean Prod., 2020, 250, 119510 CrossRef CAS.
- S. Sharma, K. Raj and S. Singh, Neurotoxic. Res., 2019, 37, 198–209 CrossRef.
- C. Wang, Z. Cai, W. Wang, M. Wei, D. Kou, T. Li, Z. Yang, H. Guo, W. Le and S. Li, J. Nutr. Biochem., 2019, 70, 147–155 CrossRef CAS.
- J. Wattanathorn, P. Chonpathompikunlert, S. Muchimapura, A. Priprem and O. Tankamnerdthai, Food Chem. Toxicol., 2008, 46, 3106–3110 CrossRef CAS PubMed.
- Q. Q. Mao, Z. Huang, S. P. Ip, Y. F. Xian and C. T. Che, Neurosci. Lett., 2011, 504, 181–184 CrossRef CAS PubMed.
- Q. Q. Mao, Y. F. Xian, S. P. Ip and C. T. Che, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2011, 35, 1144–1147 CrossRef CAS.
- S. Pattanaik, D. Hota, S. Prabhakar, P. Kharbanda and P. Pandhi, Phytother. Res., 2009, 23, 1281–1286 CrossRef CAS.
- T. Ren, M. Yang, M. Xiao, J. Zhu, W. Xie and Z. Zuo, Life Sci., 2019, 218, 314–323 CrossRef CAS PubMed.
- J. J. Hulce, A. B. Cognetta, M. J. Niphakis, S. E. Tully and B. F. Cravatt, Nat. Methods, 2013, 10, 259–264 CrossRef PubMed.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681–708 RSC.
- J. Ge, C. W. Zhang, X. W. Ng, B. Peng, S. Pan, S. Du, D. Wang, L. Li, K. L. Lim, T. Wohland and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 4933–4937 CrossRef CAS PubMed.
- K. Lee, H. S. Ban, R. Naik, Y. S. Hong, S. Son, B. K. Kim, Y. Xia, K. B. Song, H. S. Lee and M. Won, Angew. Chem., Int. Ed., 2013, 52, 10286–10289 CrossRef CAS PubMed.
- H. Guo and Z. Li, MedChemComm, 2017, 8, 1585–1591 RSC.
- S. Chen, C. Liang, H. Li, W. Yu, M. Prothiwa, D. Kopczynski, S. Loroch, M. Fransen and S. H. L. Verhelst, ACS Chem. Biol., 2023, 18, 686–692 CrossRef CAS PubMed.
- M. Soethoudt, G. Alachouzos, E. J. van Rooden, M. D. Moya-Garzon, R. van den Berg, L. H. Heitman and M. van der Stelt, Cannabis Cannabinoid Res., 2018, 3, 136–151 CrossRef CAS.
- J. Jiang, Y. Liu, S. Yang, H. Peng, J. Liu, Y. X. Cheng and N. Li, ACS Med. Chem. Lett., 2021, 12, 1905–1911 CrossRef CAS PubMed.
- R. A. Homan, A. M. Jadhav, L. P. Conway and C. G. Parker, J. Am. Chem. Soc., 2022, 144, 15013–15019 CrossRef CAS PubMed.
- T. Y. Zhu, X. T. Wu, C. Chen, X. Q. Liu, L. Zhu, J. G. Luo and L. Y. Kong, ACS Med. Chem. Lett., 2022, 13, 449–456 CrossRef CAS PubMed.
- A. H. Tantawy, M. F. El-Behairy, W. H. Abd-Allah, H. Jiang, M.-Q. Wang and A. A. Marzouk, J. Med. Chem., 2021, 64, 17468–17485 CrossRef CAS.
- S. Zhuang, Q. Li, L. Cai, C. Wang and X. Lei, ACS Cent. Sci., 2017, 3, 501–509 CrossRef CAS PubMed.
- A. Schöffmann, L. Wimmer, D. Goldmann, S. Khom, J. Hintersteiner, I. Baburin, T. Schwarz, M. Hintersteininger, P. Pakfeifer, M. Oufir, M. Hamburger, T. Erker, G. F. Ecker, M. D. Mihovilovic and S. Hering, J. Med. Chem., 2014, 57, 5602–5619 CrossRef.
- M. T. Islam, J. Hasan, H. M. S. H. Snigdha, E. S. Ali, J. Sharifi-Rad, M. Martorell and M. S. Mubarak, J. Ethnopharmacol., 2020, 257, 112853 CrossRef CAS.
- S. Li, M. Lv and H. Xu, Mini-Rev. Med. Chem., 2022, 23, 917–940 CrossRef PubMed.
- A. H. Tantawy, X.-G. Meng, A. A. Marzouk, A. Fouad, A. H. Abdelazeem, B. G. M. Youssif, H. Jiang and M.-Q. Wang, RSC Adv., 2021, 11, 25738–25751 RSC.
- A. H. Tantawy, S. M. Farag, L. Hegazy, H. Jiang and M.-Q. Wang, Bioorg. Chem., 2020, 94, 103464 CrossRef CAS PubMed.
- Y. Su, J. Ge, B. Zhu, Y.-G. Zheng, Q. Zhu and S. Q. Yao, Curr. Opin. Chem. Biol., 2013, 17, 768–775 CrossRef CAS PubMed.
- Z. Li, D. Wang, L. Li, S. Pan, Z. Na, C. Y. J. Tan and S. Q. Yao, J. Am. Chem. Soc., 2014, 136, 9990–9998 CrossRef CAS PubMed.
- H.-G. Lee, M. A. Wheeler and F. J. Quintana, Nat. Rev. Drug Discovery, 2022, 21, 339–358 CrossRef CAS.
- S. Wang, B. Wang, D. Shang, K. Zhang, X. Yan and X. Zhang, Front. Physiol., 2022, 13, 814285 CrossRef.
- Y. Song, C. Cao, Q. Xu, S. Gu, F. Wang, X. Huang, S. Xu, E. Wu and J. H. Huang, Front. Neurol., 2020, 11, 431 CrossRef PubMed.
- H. A. Burgess, T. Afrikanova, A.-S. K. Serruys, O. E. M. Buenafe, R. Clinckers, I. Smolders, P. A. M. de Witte, A. D. Crawford and C. V. Esguerra, PLoS One, 2013, 8, e54166 CrossRef.
- J. Luo, Biomedicines, 2022, 10, 1206 CrossRef CAS.
- A. Moulin, M. Mathieu, C. Lawrence, R. Bigelow, M. Levine, C. Hamel, J. P. Marquette, J. Le Parc, C. Loux, P. Ferrari, C. Capdevila, J. Dumas, B. Dumas, A. Rak, J. Bird, H. Qiu, C. Q. Pan, T. Edmunds and R. R. Wei, Protein Sci., 2014, 23, 1698–1707 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31, 455–461 CrossRef PubMed.
- J.-W. Yu, S. Li, L.-D. Bao and L. Wang, Chin. J. Nat. Med., 2021, 19, 412–421 CAS.
- A. M. Abdelhamid, A. Selim and M. A. Zaafan, Front. Mol. Biosci., 2021, 8, 754098 CrossRef CAS PubMed.
- P. Xiaoxiao, G. Xiaoqian, P. Lili and L. Liyu, Cell. Mol. Biol., 2023, 69, 239–243 CrossRef.
- D. Khan, P. Bedner, J. Müller, F. Lülsberg, L. Henning, M. Prinz, C. Steinhäuser and S. Muhammad, Mol. Neurobiol., 2023, 60, 3413–3422 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04330h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |