
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmonic circular dichroism-based metal ion detection using gold nanorod–DNA complexes†
Yali
Shi‡
a,
Satoshi
Nakamura‡
 *bc,
Hideyuki
Mitomo
bd,
Yusuke
Yonamine
b,
Guoqing
Wang
e and
Kuniharu
Ijiro
*b
*bc,
Hideyuki
Mitomo
bd,
Yusuke
Yonamine
b,
Guoqing
Wang
e and
Kuniharu
Ijiro
*b
aGraduate School of Life Science, Hokkaido University, Kita 10, Nishi 8, Kita-Ku, Sapporo 060-0810, Japan
bResearch Institute for Electronic Science, Hokkaido University, Kita 21, Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: ijiro@es.hokudai.ac.jp
cInstitute for the Promotion of General Graduate Education (IPGE), Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata 992-8510, Japan. E-mail: s.nakamura@yz.yamagata-u.ac.jp
dInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Sendai, 980-8577, Japan
eMOE Key Laboratory of Evolution and Marine Biodiversity and Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao 266003, China
First published on 27th September 2024
Abstract
We report that complexes formed between gold nanorods (AuNRs) and metal-mediated DNA exhibit plasmonic circular dichroism (CD) signals up to ∼400 times stronger than the molecular CD signal of DNA. This substantial enhancement enables the detection of metal ions, offering a promising approach to analytical applications in chiral biochemistry.
DNA has several advantages that make it an excellent biosensing probe.1–3 First, by designing the base sequence, DNA can recognize various target substances such as metal ions,4 small molecules5,6 and proteins.7 Second, DNA can undergo conformational changes upon recognition of a target substance.8 By taking advantage of these features, a variety of methods have been developed for the detection of target substances.1–3 For example, the analyte-triggered oligodeoxynucleotide (ODN) conformational change can be converted into an optical or electrical signal by modification with fluorophores9 or electroactive moieties.10 Plasmonic nanoparticles, such as gold nanoparticles, are also a powerful tool for converting ODN conformational changes into an optical signal.11 Due to localized surface plasmon resonance (LSPR), plasmonic nanoparticles exhibit a color change in accordance with the distance between the particles.12 This color change can be triggered by probe ODNs on the particle surface for detection of target substances.13 In addition, as DNA is a chiral molecule, its conformational changes can be directly detected with a circular dichroism (CD) spectrometer,14 although CD spectroscopy suffers from low detection sensitivity. For example, for a 20-base pair double-stranded (ds)ODN, a high sample concentration of ∼2 μM is required to obtain a CD signal.14 Thus, CD spectroscopy has not been widely used for DNA-based biosensors.
Induced plasmonic chirality, a phenomenon in which an achiral plasmonic nanostructure becomes optically active in response to chiral molecules,15,16 may help overcome this shortcoming. It is known that plasmonic CD is induced by dipole–dipole interactions between chiral adsorbates and plasmon nanostructures and/or by the chiral assembly of plasmonic nanoparticles.16 One notable aspect of this phenomenon is that the intensity of the plasmonic CD is much higher than that of the molecular CD.17–20 For example, Kneer et al. sandwiched a DNA origami sheet between two gold nanorods (AuNRs) and demonstrated the emergence of plasmonic CD signals ∼300 times more intense than that of the DNA origami.19 Chiral self-assemblies of AuNRs induced by helical polymers showed a strong CD response in the vis-NIR region.21 Serum albumin and AuNR complexes provided strong plasmonic CD16,22–24 and chiral assemblies of AuNRs as revealed by cryo-transmission electron microscopy (TEM).16 However, the relationship between the protein structure and plasmonic CD of AuNR–protein complexes remains unclear due to the complex protein structures.24 Based on our previous studies of plasmonic AuNR–DNA complexes,25–28 DNA can be utilized as an ideal molecule for the construction of nanoparticle–molecule complexes to induce plasmonic CD, owing to the clearer structural information. By leveraging these attributes, not only are the potential applications of such complexes expanded in terms of chiral sensing, but it also paves the way for deeper explorations into chiroptical mechanisms in nanoparticle–molecule complexes.
Although the molecular structure required to induce plasmonic chirality remains controversial, a common feature of many of these molecules is a helical structure.15–17,19,21–24,27,29,30 It is well known that Hg2+, which is highly toxic to the human body, mediates thymine mismatching and forms thymine–Hg2+–thymine (T–HgII–T) complexes.31,32 Thus, single-stranded (ss)ODN can be hybridized to form dsDNA (helical structure) in the presence of Hg2+. Additionally, thymine-rich ssDNA adopts a random coil structure (non-helical structure) due to weak base pair stacking interactions.33 Based on this insight, in this study, we demonstrate that the plasmonic CD induced by AuNR–DNA complexes can be used for metal ion detection (Scheme 1).
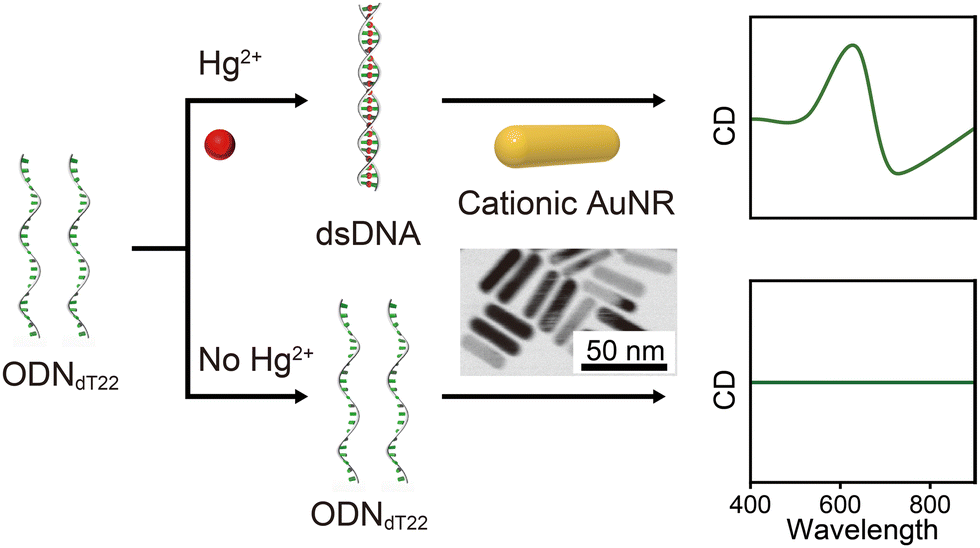 | ||
| Scheme 1 Schematic illustration of this study. The AuNR–DNA complexes, showing plasmonic CD signals based on DNA conformational changes from random coil to helix, can be used for the detection of Hg2+. | ||
First, we confirmed the structural changes in ODNdT22 (sequence: 5′-TTTTTTTTTTTTTTTTTTTTTT-3′) through recognition of Hg2+. We used Tris–HCl solutions for all experiments in this study. We measured the fluorescence intensity of a mixture of ODNdT22 (final concentration = 0.6 μM) and SYBR Green I (SG) containing Hg2+ and other metal ions (Ag+, Cr6+, Cu2+, Fe3+, and Zn2+). Note that the concentration of metal ions was set to the minimum amount (6.6 μM) required for Hg2+ to mediate all of the thymine mismatches. SG stains dsDNA more efficiently than ssDNA;34 therefore, by comparing fluorescence intensities, it is possible to confirm whether ODNdT22 adopts a ss or ds conformation. As expected, the fluorescence intensity of SG with Hg2+ was significantly greater than that for other metal ions or blank samples without Hg2+ (Fig. S1, ESI†). These results confirmed the ds formation of ODNdT22 by Hg2+ even under our experimental conditions.
Next, we investigated the formation of DNA complexes with AuNRs via electrostatic interactions. We prepared cationic AuNRs (size: ∼40 nm × ∼10 nm, zeta potential: +13.8 mV, final concentration = 16 nM) by the surface exchange of cetyltrimethylammonium bromide (CTAB)-covered AuNRs with primary amine-terminated hexaethylene glycol thiol ligands, to make them both more stable and controllable. Then, the cationic AuNRs were added into the Tris–HCl buffer solution containing ODNdT22 to form AuNR–DNA complexes. Dynamic light scattering (DLS) was used to characterize the complexes. As shown in Fig. 1A, AuNR alone showed DLS peaks at ∼1 nm and ∼39 nm. The former results from rotational motion and the latter results from translational motion.35 The mixture of AuNRs and ODNdT22 showed a DLS peak at ∼150 nm, indicating complex formation. The position of this peak was almost completely independent of the Hg2+ concentration. We then measured the extinction spectra of AuNR alone and the AuNR–ODNdT22 complexes (Fig. 1B). AuNR alone showed a transverse (T)-LSPR peak at ∼510 nm and a longitudinal (L)-LSPR peak at ∼760 nm. In contrast, AuNR–ODNdT22 complexes exhibited a T-LSPR peak around ∼530 nm and a L-LSPR peak around ∼660 nm, regardless of the presence or absence of Hg2+. The shift in the L-LSPR peak to a shorter wavelength indicates that the AuNRs assembled in a side-by-side manner.36,37 This was also supported by scanning transmission electron microscopy (STEM) imaging (Fig. 1C and D). The above DLS and extinction spectra results demonstrated the successful formation of AuNR–DNA complexes.
 | ||
| Fig. 1 (A) DLS profiles of AuNR alone (black line) and AuNR–ODNdT22 complexes prepared in the absence (blue line) or presence (red line) of Hg2+. (B) Extinction spectra of AuNR alone (black line), and AuNR–ODNdT22 complexes prepared in the absence (blue line) or presence (red line) of Hg2+. (C) and (D) Typical STEM images of the AuNR–ODNdT22 complexes prepared in the absence (C) and presence (D) of Hg2+. (AuNRs: 16 nM; ODNdT22: 0.6 μM; Hg2+: 6.6 μM). | ||
Although the size and morphology of the AuNRs assemblies did not differ markedly (Fig. 1), there was a clear difference between the optical activities of the AuNR–ODNdT22 complexes with and without Hg2+. Fig. 2A shows their CD spectra. Note that all CD spectra shown in this work have been smoothed using the Savitzky–Golay method (Fig. S2, ESI†).38 In the absence of Hg2+, neither AuNR alone nor the AuNR–ODNdT22 complexes showed plasmonic CD in the visible region (Fig. 2A). In contrast, the AuNR–ODNdT22 complexes with Hg2+ showed negative couplet CD with a dip at ∼720 nm and a peak at ∼620 nm. In the UV region, the AuNR–ODNdT22 complexes with Hg2+ exhibit similar but distinct CD characteristics to those without AuNRs in the same region due to the large electromagnetic field near the Hg2+-mediated DNA.39 Indeed, similar results were observed in the AuNR–dsDNA complexes in which dsDNA was formed by ODNdT22 and ODNdA22 (sequence: 5′-AAAAAAAAAAAAAAAAAAAAAA-3′) (Fig. S3A, ESI†).
 | ||
| Fig. 2 CD spectra for (A) 16 nM AuNR alone (black line), AuNR and 0.6 μM ODNdT22 complexes (blue line), AuNR and ODNdT22 complexes with 6.6 μM Hg2+ (red line), (B) 20 μM ODNdT22 alone (blue line) and with 220 μM Hg2+ (red line). | ||
Here we emphasize three important characteristics. First, the plasmonic CD was much larger than the molecular CD of the DNA in the UV region (the plasmonic CD was detected even though the DNA was below the detection limit). To quantitatively compare the plasmonic CD with the CD of the DNA, we used the following equation:17,20

In order to test the analytical performance with regard to metal ion detection using the AuNR–DNA complexes, the plasmonic chiroptical activities of the AuNR–ODNdT22 complexes were recorded upon addition of Hg2+ at different concentrations (Fig. 3A). Even when the Hg2+ concentration was changed, the zero-crossing (∼670 nm) and peak (∼610 nm) positions remained nearly identical. In addition, as shown in Fig. S4 (ESI†), the zero-crossing and the L-LSPR peak were well matched. Fig. 3B plots the ΔCD(plasmon)versus Hg2+ concentration, which shows that the plasmonic CD began to be detected around the Hg2+ concentration of 2 μM. This is consistent with the Hg2+ concentration at which ODNdT22 began to form double strands (Fig. S1B, ESI†). When the concentration of Hg2+ was over 6.6 μM, ΔCD reached its largest value (∼25 mdeg). The saturation may be explained by the fact that ssODNdT22 was completely consumed in the presence of 6.6 μM of Hg2+. It is also worth noting that the plasmonic CD signal in our system did not decrease, as in the fluorescence method, in the presence of high Hg2+ concentrations (Fig. S1, ESI†), indicating its good stability with regard to Hg2+ detection. We also tested the plasmonic CD responses to the other potential interfering compounds, including Ag+, Cr6+, Cu2+, Fe3+, and Zn2+, under the same reaction conditions. Interestingly, except for Hg2+ with reproducibility (Fig. S5, ESI†), no obvious plasmonic CD signal was obtained in the presence of the other interfering ions (6.6 μM) in the sensing systems, indicating good selectivity toward Hg2+ (Fig. 3B). These results indicated that the AuNR–DNA complex system shows high analytical performance with regard to metal ions.
 | ||
| Fig. 3 (A) CD spectra for AuNR–ODNdT22 complexes prepared at different concentrations of Hg2+. (B) Plots of ΔCD versus Hg2+ or other metal ion concentrations (6.6 μM). | ||
The advantage of our detection method is that the ion of interest can be changed simply by changing the base sequence. To demonstrate this, we performed an experiment using Ag+ as another model ion. For this experiment, a probe ODN with the sequence 5′-CCCCCCCCCCCCCCCCCCCCCC-3′ (ODNdC22) was used (Fig. 4A). ODNdC22 underwent a conformation change caused by Ag+ as Ag+ mediates cytosine mismatching and formed cytosine–Ag+–cytosine (C–AgI–C) base pairs,40 as confirmed by CD measurements (Fig. 4B). As shown in Fig. 4A, the AuNR–ODNdC22 complexes showed no plasmonic CD in the absence of Ag+. However, in the presence of Ag+, the AuNR–ODNdC22 complexes exhibit negative couplet plasmonic CD. The ACD from AuNR–ODNdC22 complexes at Ag+ concentration of 6.6 μM was calculated to be 400. It is noteworthy that the AuNR–ODNdC22 complexes exhibit a marked plasmonic CD. Specifically, AuNR–ODNdC22 complexes with Ag+ (6.6 μM) show a higher value of g-factor (1.0 × 10−2), which represents the dissymmetry, whereas the AuNR–ODNdT22 complex with Hg2+ (6.6 μM) shows a lower value (0.09 × 10−2) (Fig. S6) (details are in the ESI†). This value is comparable to that of the AuNR–protein complex system, which has side-by-side AuNR chiral assemblies (1.9 × 10−2).23
 | ||
| Fig. 4 CD spectra for (A) 16 nM AuNR alone (black line), AuNR and 0.6 μM ODNdC22 complexes (blue line), AuNR and ODNdC22 complexes with 6.6 μM Ag+ (red line), (B) 20 μM ODNdC22 alone (blue line) and with 220 μM Ag+ (red line). | ||
In summary, we have demonstrated that conformational changes in DNA can be observed by the induced plasmonic optical activity from AuNR–DNA complexes, which has afforded the ability to detect metal ions (Hg2+ and Ag+) with no significant background signal. Importantly, the induced plasmonic CD in the visible region was much larger than the molecular CD of the ODN in the UV region. It is also noteworthy that clear changes in plasmonic CD could be caused even with metal-mediated ODNs that show little molecular CD change. We believe that this study will inspire further research on chiral plasmonic applications and facilitate exploration into the chiroptical mechanisms.
We acknowledge financial support from the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant no. 24K03258 and 24K17574). S. N. acknowledges financial support from Iketani Science and Technology Foundation. K. I. acknowledges support from the “Photoexcitonix Project” and the “Project of Young Investigator Promotion” at Hokkaido University. This work was also supported in part by the Crossover Alliance to Create the Future with People, Intelligence and Materials” from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT). A part of this work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT): Grant Number JPMXP1223HK0051 and JPMXP1224HK0048 (Hokkaido University).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Tjong, L. Tang, S. Zauscher and A. Chilkoti, Chem. Soc. Rev., 2014, 43, 1612–1626 RSC.
- H.-M. Meng, H. Liu, H. Kuai, R. Peng, L. Mo and X.-B. Zhang, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- H. Yoo, H. Jo and S. S. Oh, Mater. Adv., 2020, 1, 2663–2687 RSC.
- M. Rajendran and A. D. Ellington, Anal. Bioanal. Chem., 2008, 390, 1067–1075 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850–852 CrossRef CAS PubMed.
- D. E. Huizenga and J. W. Szostak, Biochemistry, 1995, 34, 656–665 CrossRef CAS PubMed.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS PubMed.
- R. Nutiu and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 1061–1065 CrossRef CAS PubMed.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- C. Fan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9134–9137 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- L. Wang, Z. He, Q. Chen, G. Wang, X. Liang, T. Takarada and M. Maeda, ACS Sustainable Chem. Eng., 2023, 11, 3611–3620 CrossRef CAS.
- J. Kypr, I. Kejnovská, D. Renčiuk and M. Vorlíčková, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- B. M. Maoz, Y. Chaikin, A. B. Tesler, O. B. Elli, Z. Fan, A. O. Govorov and G. Markovich, Nano Lett., 2013, 13, 1203–1209 CrossRef CAS PubMed.
- Q. Zhang, T. Hernandez, K. W. Smith, S. A. H. Jebeli, A. X. Dai, L. Warning, R. Baiyasi, L. A. McCarthy, H. Guo, D.-H. Chen, J. A. Dionne, C. F. Landes and S. Link, Science, 2019, 365, 1475–1478 CrossRef CAS PubMed.
- F. Lu, Y. Tian, M. Liu, D. Su, H. Zhang, A. O. Govorov and O. Gang, Nano Lett., 2013, 13, 3145–3151 CrossRef CAS PubMed.
- R.-Y. Wang, P. Wang, Y. Liu, W. Zhao, D. Zhai, X. Hong, Y. Ji, X. Wu, F. Wang, D. Zhang, W. Zhang, R. Liu and X. Zhang, J. Phys. Chem. C, 2014, 118, 9690–9695 CrossRef CAS.
- L. M. Kneer, E.-M. Roller, L. V. Besteiro, R. Schreiber, A. O. Govorov and T. Liedl, ACS Nano, 2018, 12, 9110–9115 CrossRef CAS PubMed.
- W. Wang, F. Wu, Y. Zhang, W. Wei, W. Niu and G. Xu, Chem. Commun., 2021, 57, 7390–7393 RSC.
- G. Cheng, D. Xu, Z. Lu and K. Liu, ACS Nano, 2019, 13, 1479–1489 CAS.
- S. Hou, H. Zhang, J. Yan, Y. Ji, T. Wen, W. Liu, Z. Hu and X. Wu, Phys. Chem. Chem. Phys., 2015, 17, 8187–8193 RSC.
- H. Shinmori and C. Mochizuki, Chem. Commun., 2017, 53, 6569–6572 RSC.
- L. A. Warning, A. R. Miandashti, A. Misiura, C. F. Landes and S. Link, J. Phys. Chem. C, 2022, 126, 2656–2668 CrossRef CAS.
- S. Nakamura, H. Mitomo, M. Aizawa, T. Tani, Y. Matsuo, K. Niikura, A. Pike, M. Naya, A. Shishido and K. Ijiro, ACS Omega, 2017, 2, 2208–2213 CrossRef CAS PubMed.
- S. Nakamura, H. Mitomo, Y. Sekizawa, T. Higuchi, Y. Matsuo, H. Jinnai and K. Ijiro, Langmuir, 2020, 36, 3590–3599 CrossRef CAS PubMed.
- S. Nakamura, H. Mitomo, Y. Yonamine and K. Ijiro, Chem. Lett., 2020, 49, 749–752 CrossRef.
- S. Nakamura, H. Mitomo, S. Suzuki, Y. Torii, Y. Sekizawa, Y. Yonamine and K. Ijiro, Chem. Lett., 2022, 51, 529–532 CrossRef CAS.
- Y. Zhu, L. Xu, W. Ma, Z. Xu, H. Kuang, L. Wang and C. Xu, Chem. Commun., 2012, 48, 11889–11891 RSC.
- W. Ma, H. Kuang, L. Xu, L. Ding, C. Xu, L. Wang and N. A. Kotov, Nat. Commun., 2013, 4, 2689 CrossRef PubMed.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172–2173 CrossRef CAS PubMed.
- L. Benda, M. Straka, V. Sychrovský, P. Bouř and Y. Tanaka, J. Phys. Chem. A, 2012, 116, 8313–8320 CrossRef CAS PubMed.
- W. Saenger, Principles of Nucleic Acid Structure, Springer New York, New York, NY, 1984 Search PubMed.
- H. Zipper, H. Brunner, J. Bernhagen and F. Vitzthum, Nucleic Acids Res., 2004, 32, e103 CrossRef PubMed.
- M. Glidden and M. Muschol, J. Phys. Chem. C, 2012, 116, 8128–8137 CrossRef CAS.
- M. Gluodenis and C. A. Foss, J. Phys. Chem. B, 2002, 106, 9484–9489 CrossRef CAS.
- J. Yang, Y. Sekizawa, X. Shi, K. Ijiro and H. Mitomo, Bull. Chem. Soc. Jpn., 2024, 97, uoae073 CrossRef.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- Z. Zhu, W. Liu, Z. Li, B. Han, Y. Zhou, Y. Gao and Z. Tang, ACS Nano, 2012, 6, 2326–2332 CrossRef CAS PubMed.
- A. Ono, S. Cao, H. Togashi, M. Tashiro, T. Fujimoto, T. Machinami, S. Oda, Y. Miyake, I. Okamoto and Y. Tanaka, Chem. Commun., 2008, 4825–4827 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04017a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |