
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanically interlocked host systems for ion-pair recognition†
Arya
Arun
ab,
Hui Min
Tay
 a and
Paul D.
Beer
*a
a and
Paul D.
Beer
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
bDepartment of Chemistry, University of Oxford, Rodney Porter Building, Sibthorp Road, Oxford OX1 3QU, UK
First published on 16th September 2024
Abstract
The ever-increasing interest directed towards the construction of host architectures capable of the strong and selective recognition of various ionic species of biological, medical and environmental importance has identified mechanically interlocked molecules (MIMs), such as rotaxanes and catenanes, as potent host systems, owing to their unique three-dimensional topologically preorganised cavity recognition environments. Ion-pair receptors are steadily gaining prominence over monotopic receptor analogues due to their enhanced binding strength and selectivity, demonstrated primarily through acyclic and macrocyclic heteroditopic host systems. Exploiting the mechanical bond for ion-pair recognition through the strategic design of neutral heteroditopic MIMs offers exciting opportunities to accomplish potent and effective binding while mitigating competing interactions from the bulk solvent and counter-ions. This review details the design and ion-pair recognition capabilities of rotaxanes and catenanes employing hydrogen bonding (HB) and halogen bonding (XB) motifs, providing valuable insight into the burgeoning field and inspiration for future research.
 Arya Arun | Arya Arun obtained her MSc (Hons.) in Chemistry from Birla Institute of Technology and Science, Pilani in 2019, following which she completed her DPhil in Inorganic Chemistry at the University of Oxford under the supervision of Professor Paul D. Beer. Her research in the Beer group focused on the synthesis of mechanically interlocked molecules (MIMs) for applications in host–guest recognition and salt-extraction. Currently, she is working as a Postdoctoral Research Associate at the University of Oxford with Dr Patrick Rabe on the design and synthesis of metal photocages and substrate analogues for bioluminescent enzymes. |
 Hui Min Tay | Hui Min Tay completed a Bachelor of Science (Honours) at the University of Melbourne in 2020, working under the supervision of Prof. Brendan Abrahams and Dr Carol Hua to develop chiral metal–organic frameworks for enantiomeric discrimination. She is now completing a DPhil in Inorganic Chemistry at the University of Oxford under the supervision of Professor Paul D. Beer. Her research interests lie in the design and synthesis of mechanically interlocked host systems for recognition and sensing of charged guests. |
 Paul D. Beer | Paul Beer obtained a PhD from King's College London in 1982 with Dr C. Dennis Hall. After a Royal Society European Post-doctoral Fellowship with Professor J.-M. Lehn and a Demonstratorship at the University of Exeter, he was awarded a Lectureship at the University of Birmingham in 1984. In 1990, he moved to the University of Oxford where he was made a University Lecturer and Fellow at Wadham College, and became a Professor in 1998. His research interests focus on supramolecular host–guest chemistry and coordination chemistry. |
Introduction
Driven by the prevalence and importance of charged species in chemistry,1,2 biology3–5 and the environment,6–8 the design of molecular host systems for ion recognition remains a crucial research focus within supramolecular chemistry, with ongoing efforts aimed at enhancing binding affinities and selectivity profiles through strategic structural host design.The development of supramolecular anion receptors has lagged behind its sister field of cation coordination, reflecting the greater challenges associated with anion binding, including relatively low charge densities, pH sensitivity, variable geometries and high hydration enthalpies of anions.9 Indeed, anion receptors capable of functioning in competitive aqueous-containing media have traditionally utilised multiple positively charged scaffolds.10–15 Despite the gains in anion affinity elicited by employing favourable Coulombic interactions, their non-directional nature can lead to compromised binding selectivity. Additionally, the nominally ‘non-coordinating’ counter-anions of the positively charged receptor may compete with the target anion guest. From a functional perspective, the reliance on positively charged binding motifs limits the potential applications of anion receptors; for instance, anion transport across lipid bilayers typically requires lipophilic carrier molecules.16,17 To address these limitations, interest in the design and synthesis of neutral anion receptors has been steadily growing in recent years.
In this context, mechanically interlocked molecules (MIMs) offer distinct advantages over traditional acyclic or macrocyclic hosts due to their unique structural and topological features (Fig. 1). The pre-organised and solvent-shielded cavities of MIMs present unique opportunities to design three dimensional binding sites for guest encapsulation, which through non-covalent donor/acceptor group functionalisation can be tuned to achieve size and shape complementarity towards a target guest.18–25 The resulting improvements in binding affinity and selectivity over non-interlocked receptor analogues, termed a ‘mechanical bond effect’ (MBE),26 have motivated the construction of numerous MIM-based cation27–31 and anion32–37 receptors over the years. In particular, the development of highly potent charge-neutral binding motifs has enabled the design of neutral MIM-based receptors for various novel applications, broadening the potential functionality of this class of receptors. Pioneering studies on transition metal cation template-directed synthesis of MIMs by Sauvage and co-workers demonstrated the potential of neutral catenanes to bind and stabilise low oxidation states of transition metal cations such as copper(I) via encapsulation of the guest within a bis-phenanthroline-functionalised interlocked cavity.38–44 In more recent years, Goldup and co-workers designed a series of neutral [2]rotaxanes capable of binding transition metal cations in CH3CN, wherein the confined environment of the interlocked cavity enforces unusual coordination geometries and redox state stabilisation.45
 | ||
| Fig. 1 Mechanical bond effect enhanced ion-pair recognition. | ||
Notable advancements have also been made in the area of neutral anion binding MIMs, with Beer and co-workers reporting a template-directed synthetic strategy driven by potent halogen bonding (XB)⋯Cl− interactions to prepare neutral interlocked hosts capable of halide anion recognition in aqueous–acetone solvent mixtures.46 This strategy was recently employed in the preparation of the first hetero[2]catenanes capable of binding and selectively transporting halide anions across a phospholipid bilayer.47 The compact geometry and lipophilic nature of the catenane hosts enabled facile membrane transversal while their strong binding affinity and preference for halides translated to an impressive transport selectivity for chloride over nitrate and hydroxide anions. Rotaxane hosts prepared using a similar template-directed approach have also shown promise as selective colorimetric sensors for chloride anions.48 Recently, Chmielewski and co-workers reported a charge–neutral homo[2]catenane synthesised via a novel sulfate anion template-directed strategy, which showed impressive binding affinity and selectivity for sulfate over other oxoanions and halides, and was capable of functioning as a sensitive fluorescence sensor for sulfate anions in competitive 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMSO/H2O solvent mixtures.49
:1 DMSO/H2O solvent mixtures.49
While charge–neutral MIM-based receptors are increasingly finding applications in cation and anion recognition, their greatest advantage over charged receptors lies in their propensity to be utilised as heteroditopic receptors capable of simultaneously binding a cation and anion ion-pair. Ion-pair recognition has emerged as a powerful strategy to augment the binding properties of a receptor by exploiting favourable proximal cation–anion electrostatic interactions and, in some cases, conformational allosteric cooperativity associated with binding oppositely charged guests.50 Indeed, studies conducted on acyclic/macrocyclic heteroditopic ion-pair receptors generally reveal improved binding affinities for ion-pairs or constituent ions relative to monotopic receptor analogues.51 This has encouraged their use in a myriad of applications, including salt extraction/solubilisation,52–62 membrane transport63,64 and the recognition of biologically-relevant zwitterions.65,66
Mechanically interlocked molecules are promising scaffolds upon which cation and anion binding sites can be incorporated in a modular fashion to construct complex heteroditopic receptors. While still in its infancy, the field has gained significant traction in recent years, with a growing diversity of catenane- and rotaxane-based heteroditopic hosts that show promising ion-pair binding properties and potential applications in ion-pair sensing and extraction.67–72
In this article, we present prominent developments in the field of mechanically interlocked heteroditopic host molecules for ion-pair recognition, ranging from early receptors that utilise traditional hydrogen bonding (HB) interactions for anion binding to recent examples featuring halogen bonding (XB) donor groups. In particular, we highlight the importance of the interlocked receptor topology in creating solvent–shielded binding cavities and its role in dictating MBE enhanced ion-pair affinity and selectivity profiles of the receptors (Fig. 1).
Heteroditopic MIM-based receptors
Heteroditopic receptors typically consist of one or more cation and anion binding sites, which can be located in close proximity to each other or spatially separated. This forms the basis for a broad general classification of ion-pair receptors into contact ion-pair or separated ion-pair receptors (Fig. 2). Contact ion-pairs are formed when the two bound ions are held proximal to each other within the receptor, as determined by X-ray crystallographic studies. In contrast, when the cation and anion binding sites are distal to each other, the co-bound ion-pair may be separated by solvent molecules (solvent-separated) or the receptor framework (host-separated).73 | ||
| Fig. 2 Ion-pair binding modes in heteroditopic receptors: (a) host-separated; (b) solvent-separated; (c) direct contact. | ||
The literature surrounding supramolecular ion-pair recognition has been largely dominated by reports of heteroditopic receptors for alkali metal halide binding,52,61,63,64,74–86 owing to the ubiquity of these salts across a range of biological and environmental systems. For instance, sodium and potassium cations are crucial in maintaining cellular electrolyte concentrations, while the mis-regulation of chloride and iodide in the body has been associated with various diseases.87–89 Recently, methods for detection and recovery of lithium salts have grown in prominence with the large-scale development of lithium-ion battery technologies.90,91 Heteroditopic receptors capable of binding alkali metal halide ion-pair binding typically utilise hard oxygen donors for metal cation coordination, often introduced as crown ether or poly(ethylene glycol) moieties, alongside a range of neutral hydrogen bonding, and more recently halogen bonding, donor groups for halide binding. The synthetic accessibility of these binding motifs, as well as their proven ability for strong and selective alkali metal halide binding, have encouraged their integration into a range of MIM-based heteroditopic receptors.
MIM heteroditopic receptors utilising hydrogen bonding (HB) interactions
Despite the advantages of their unique topologies and preorganised interlocked cavities that have in particular been shown to enhance anion guest recognition capabilities, examples of neutral heteroditopic MIMs remain relatively scarce. Early examples of neutral hydrogen bonding interlocked frameworks capable of ion-pair recognition include Smith's [2]rotaxane 1a comprising a macrobicyclic component functionalised with a crown ether cation and isophthalamide anion binding units (Fig. 3(a)).78 The synthesis of the rotaxane 1a utilises a discrete potassium cation template bound to the dibenzo-18-crown-6 ether group to direct the association of an anionic phenolate half-axle through the isophthalamide cavity. Subsequent stoppering esterification reactions afforded the [2]rotaxane in 20% yield. The crucial role played by the potassium cation template was highlighted by the failure to form the desired rotaxane product in the absence of the potassium cation as well as in the presence of a larger cesium cation. 1H-NMR anion binding studies conducted with the neutral [2]rotaxane 1a in DMSO-d6/CD3CN solvent mixtures reveal enhanced chloride anion affinities relative to the free heteroditopic macrobicycle receptor 1b (Fig. 3(b)) (Ka = 300 M−1 and Ka = 50 M−1 respectively), attributable to the steric shielding effect of the interlocked binding cavity inhibiting the competing DMSO complexation previously reported to occur within the free macrocycle 1b. Interestingly, whilst macrobicycle 1b exhibits a 7-fold enhancement in chloride binding affinity in the presence of potassium, no enhancement in chloride binding is observed in the case of the [2]rotaxane 1a. This lack of cooperativity is ascribed to the aforementioned steric effects engendered by the interpenetrating axle which disrupt the electrostatic interactions between the co-bound ions. Furthermore, 1H-NMR studies in polar organic solvents suggest that the presence of a K+ cation serves to ‘lock’ the dynamic co-conformations assumed by the components in the free state, as evidenced by the sharpening of the proton signals in the 1H-NMR spectrum. By contrast, chloride complexation fails to disrupt the dynamic inter-component motions likely due to the lack of any axle–anion interactions. | ||
| Fig. 3 (a) Smith's heteroditopic [2]rotaxane 1a capable of KCl ion-pair binding; (b) macrobicyclic precursor 1b, which previously also demonstrated cooperative binding of KCl ion-pairs. | ||
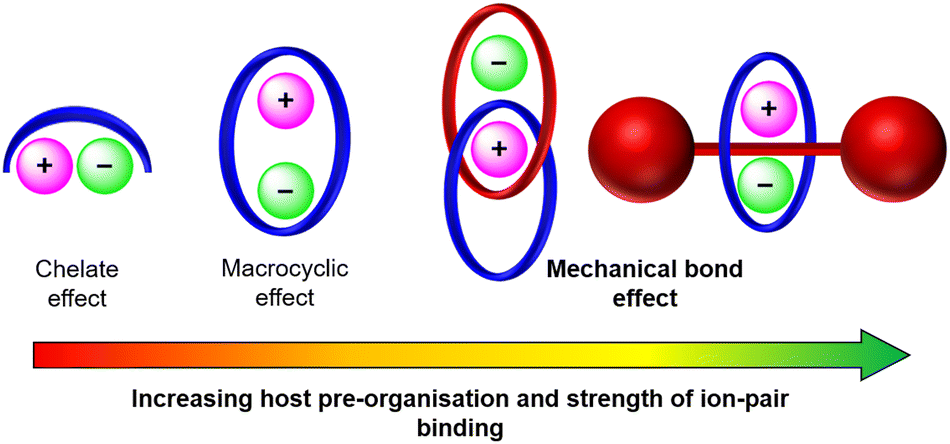
Since then only a handful of neutral hydrogen bond donor containing heteroditopic MIMs have been reported, including Beer's neutral [2]rotaxane 2 consisting of a pyridine N-oxide axle and an isophthalamide bridged calix[4]diquinone macrocycle operating as an axle-separated ion-pair receptor (Fig. 4(a)).67 Synthesised using a sodium cation-template driven threading methodology followed by copper(I) catalysed-mediated CuAAC stoppering reactions, the resulting [2]rotaxane demonstrates remarkable alkali metal halide ion-pair recognition in 4:1 CDCl3/CD3OD solvent media. 1H-NMR based characterisation studies suggest that the free rotaxane 2 adopts a co-conformation where the axle pyridine N-oxide forms intercomponent hydrogen bonds with the macrocycle bis-amide isophthalamide group. Sodium cation complexation thereafter pre-organises the pyridine N-oxide motif to pirouette within the macrocycle towards the calixdiquinone unit. Quantitative 1H-NMR studies conducted on 2 reveal significant amplification in the halide binding constants in the presence of co-bound sodium and potassium cations, displaying a striking 15-fold cooperativity factor in the case of the NaCl ion-pair. This enhancement is accredited to favourable axle-separated electrostatic interactions between the co-bound ions and receptor preorganisation in the presence of the alkali metal cation. More recently, the same authors prepared an analogous [2]catenane 3via a sodium cation-template activated ester amide condensation clipping methodology (Fig. 4(b)), wherein the [2]catenane was serendipitously isolated with a NaCl ion-pair endotopically bound within the binding cavities separated by the macrocycles, characterised by single crystal X-ray structural analysis (Fig. 4(c)).92
 | ||
| Fig. 4 Knighton and Beer's heteroditopic (a) [2]rotaxane 2 and (b) [2]catenane 3, based on interlocked calix[4]diquinone and pyridine-N-oxide components. (c) Single crystal XRD structure of [2]catenane bound to an NaCl ion-pair. | ||
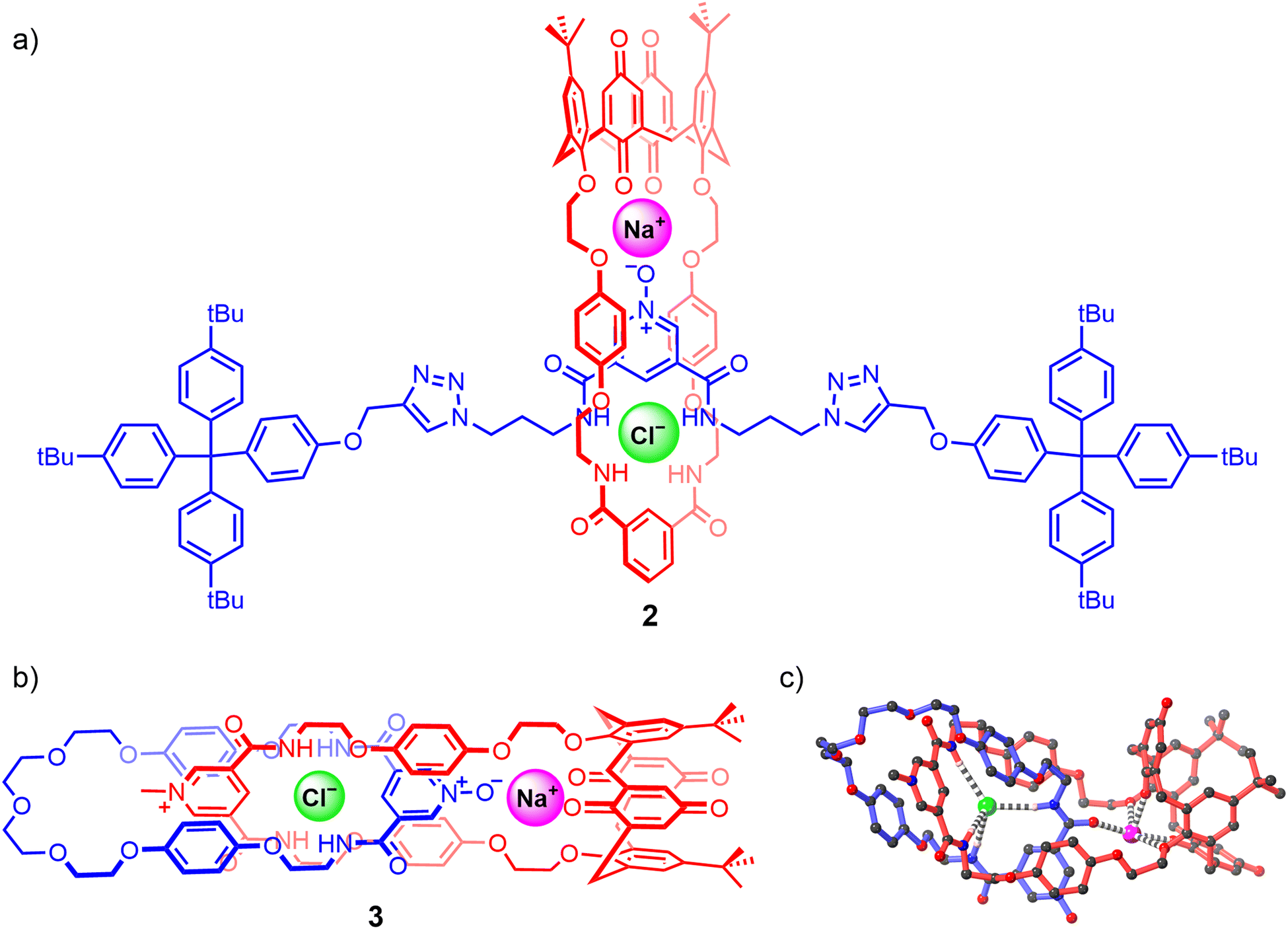
Another recent example includes Goldup's fluorescent [2]rotaxane 4 comprising a urea-based naphthalimide axle component and bipyridine functionalised macrocycle, synthesised using active metal template methodology in 92% yield (Fig. 5).71 Analysis of 1H-NMR, UV-vis and fluorescence titration data of 4 reveals that in the free state, the bipyridine group in the macrocycle interacts with the anion binding urea motif in the axle via strong HB interactions, precluding it from participating in anion binding even in the presence of highly basic anions such as AcO− and F−. By contrast, the free axle exhibits similar binding trends albeit with higher binding affinities. Notably, protonating the bipyridine motif enables macrocyclic translocation away from the urea unit in the axle, consequently enabling exogenous anion recognition at the urea station in 1:1 CDCl3/CD3CN solvent mixtures (Ka(Cl−) >104 M−1, Ka(Br−) = 4.7 × 103 M−1, Ka(HSO4−) = 2.3 × 103 M−1). Interestingly, while the protonated rotaxane 4·H+ shows a greater affinity towards less basic anions in comparison to the free axle, presumably due to favourable electrostatic interactions between the co-bound ions, addition of basic anions like AcO− and F− predictably results in deprotonation of the interlocked system.
 | ||
| Fig. 5 Goldup's dynamic [2]rotaxane shuttle 4 capable of cooperative HCl ion-pair binding. | ||
Calix[4]pyrroles and their derivatives have been formerly known to function as heteroditopic receptors, binding anions through the cone-shaped pyrrole NH groups and cations via the electron rich bowl-shaped cavity formed by the calix[4]pyrrole anion complex.93 Looking to exploit the ambidentate nature of calix[4]pyrroles, Ballester and co-workers sought to design a rigid three-dimensional macrocyclic scaffold containing two calix[4]pyrrole units, envisioned to bind ion-pairs by forming cascade complexes.94 Encouraged by the binding behaviour exhibited by the macrocycle, the authors synthesised a bis-calix[4]pyrrole functionalized [2]rotaxane 5 prepared using CuAAC ‘click’ reactions to stopper a pseudo-[2]rotaxane assembly involving a pyridine N-oxide axle thread and the bis-calix[4]pyrrole-containing cylindrical macrocycle component (Fig. 6).701H-NMR based binding studies indicated that 5 strongly bound tetraalkylammonium salts of Cl−, OCN− and NO3− in a 1:1 binding stoichiometry in CDCl3. Quantitative ITC titration experiments further revealed that the interlocked system demonstrated a preference for TBANCO (Ka = 7.9 × 105 M−1) over TBANO3 (Ka = 4 × 104 M−1) and TBACl (Ka = 5 × 104 M−1) in chloroform, likely due to host–guest size complementarity between the cylindrical cavity and linear NCO− anion. Notably, replacing the organic cation to methyltrioctylammonium (MTO+) resulted in a 20-fold increase in the binding of MTOCl relative to TBACl, presumably due to better encapsulation of the quaternary ammonium cation within the calix[4]pyrrole unit.
 | ||
| Fig. 6 Ballester's [2]rotaxane 5 for the recognition of quaternary ammonium salts. | ||
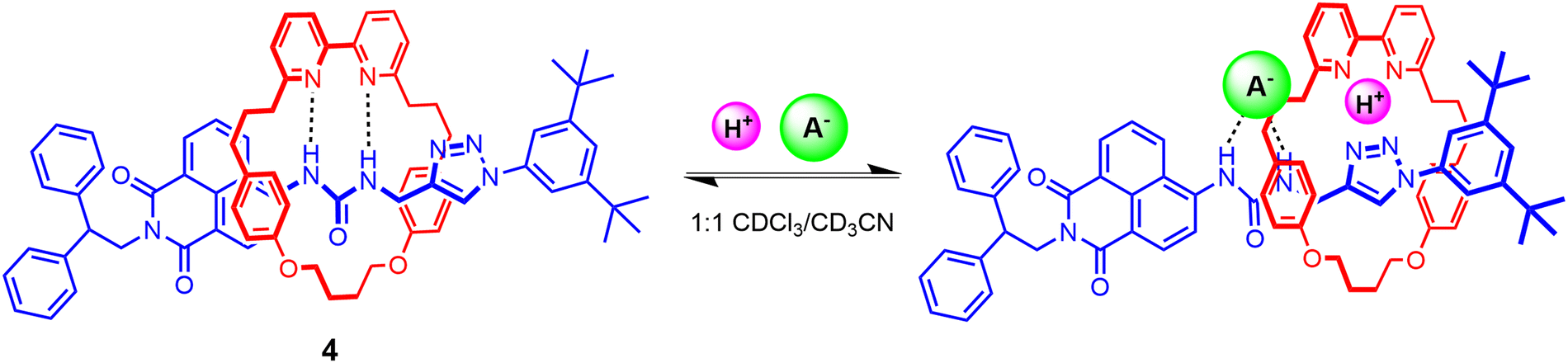
Squaramides are another well-known class of neutral, potent hydrogen bonding anion receptors. However, the unique ambidentate properties of the squaramide motif allows for the potential simultaneous binding of anions through the Lewis acidic NH donors and cations via bidentate chelation of the Lewis basic carbonyl groups. Beer and co-workers recently demonstrated for the first time ambidentate squaramide ion-pair binding behaviour through a series of HB functionalized heteroditopic [2]rotaxanes 6a–c wherein the squaramide-based axle component played a pivotal role in achieving axle-separated sodium halide ion-pair recognition (Fig. 7).95 Taking inspiration from Chiu's alkali metal template directed MIM synthesis,96 an unprecedented sodium cation-template directed strategy driven by orthogonal coordination of the sodium cation by the Lewis basic carbonyls of the axle squaramide unit and the polyether/pyridyl region of the macrocycle was employed to facilitate threading and subsequent mechanical bond formation.
 | ||
| Fig. 7 (a) Propensity of the squaramide motif for metal cation and ion-pair recognition, highlighting its ambidentate coordination mode, (b) Cartoon showing ion-pair recognition in target squaramide axle containing [2]rotaxane host systems, (c) Beer's squaramide-based [2]rotaxanes 6a–c for the recognition of sodium halide ion-pairs. | ||
In order to investigate the factors governing the ion-pair association capabilities of the interlocked squaramide axle containing [2]rotaxane hosts, macrocycles with varying cation and anion binding units were chosen, functionalized with either crown-ether like arrangements of oxygen donors or a combination of polyether and pyridyl isophthalamide linkages. 1H–1H ROESY NMR characterisation indicated that the free squaramide rotaxanes adopted co-conformations where the axle squaramide carbonyls were engaged in hydrogen bonding interactions with the bis-amide isophthalamide unit of the macrocycle in concert with analogous interactions between the macrocycle polyether/pyridyl units and axle squaramide NH groups. Notably, sodium cation binding co-conformationally pre-organises the macrocycle such that amide donors from both the macrocycle and axle form an intercomponent convergent hydrogen bond donor site for cooperative anion recognition. Extensive cation, anion and ion-pair 1H-NMR titration studies in 3:7 CD3CN/CDCl3 revealed the [2]rotaxane hosts to cooperatively bind and extract sodium halide salts, engendering up to a ca. 20-fold enhancement in bromide and iodide binding strength in the presence of an equivalent of sodium cation (Ka = 494 to 104 M−1 and Ka = 67 to 1298 M−1 respectively for the pyridyl functionalized [2]rotaxane 6c). This remarkable amplification in association constant magnitudes was attributed to favourable coulombic electrostatic interactions between the co-bound ions and co-conformational allosteric effects instigated by the ion-binding event. In the case of the NaCl ion-pair, exogenous ion-pairing salt recombination equilibrium necessitated the calculation of % bound sodium cation to the [2]rotaxane systems in order to rationalise the differing chloride binding properties of the sodium complexed receptors. While systems with macrocycles containing the crown-ether arrangement (6a) and polyether linkages (6b) failed to bind the NaCl ion-pair, the pyridyl-functionalised rotaxane 6c successfully overcame the NaCl contact ion-pair lattice enthalpy, underpinning the crucial interplay of strong concomitant cation and anion binding affinities of the receptors in enabling ion-pair recognition.
Heteroditopic receptors utilising σ-hole interactions
Although hydrogen bonding binding motifs have traditionally dominated the field of anion recognition, recently the integration of σ-hole interactions into acyclic, macrocyclic and mechanically interlocked anion receptor systems has emerged as a promising alternative strategy. In particular, halogen bonding (XB)97 interactions have grown in prominence by virtue of their often superior anion binding strength, more stringent linear interaction geometries and contrasting selectivity profiles relative to analogous HB receptors.50,98–102 Encouraged by these studies, we have begun to explore the potential of incorporating XB and related σ-hole binding motifs into heteroditopic ion-pair receptors as a means to further tune ion-pair binding affinity and selectivity.Acyclic and macrocyclic heteroditopic receptors utilising σ-hole interactions
The potential efficacy of using halogen bonding and related σ-hole interactions to modulate the affinity and selectivity profile of ion-pair receptors was initially demonstrated in non-interlocked heteroditopic host molecules. The first example of using halogen bonding interactions in ion-pair binding was reported in 2005 by Resnati and co-workers, who synthesised a tripodal heteroditopic host 7 comprising a tris(polyoxyethylene)amine cation recognition site and 4-iodotetrafluorophenyl anion recognition sites (Fig. 8(a)). 1H NMR competition studies conducted in 60:35:5 CDCl3/CD3OD/D2O showed 7 was able to bind an NaI ion-pair with significantly higher affinity than the non-XB pentafluorophenyl analogue (KXBa = 2.6 × 105 M−1; KHBa = 1.3 × 104 M−1). However, single crystal X-ray analysis suggested that only one iodine donor in 7 participates in XB interactions with the iodide guest due to the divergent arrangement of the donor motifs.103 Subsequently, Schubert and co-workers integrated a iodotriazole anion binding group into a tri(ethylene glycol)-containing macrocycle to generate a heteroditopic receptor 8XB capable of binding sodium iodide ion-pairs in 3:1 CD2Cl2/CD3CN solvent mixtures with significant positive cooperativity between the cation and anion binding events (Fig. 8(b)). In contrast, the hydrogen bonding analogue 8HB showed no measurable ion-pair binding in the same solvent system, which was attributed to intramolecular hydrogen bond formation between the prototriazole and ether oxygen atoms outcompeting guest binding.104
 | ||
| Fig. 8 Heteroditopic ion-pair receptors utilising XB and ChB interactions: (a) Resnati's tripodal receptor 7; (b) Schubert's triazole-incorporated cyclic ethylene glycol receptor 8; (c) Taylor's B15C5-functionalised XB receptor 9; (d) Docker's B15C15-functionalised ChB receptor 10. | ||
In an elegant demonstration of utilising allosteric cooperativity in receptor design, a series of potassium halide-selective acyclic ion-pair receptors were constructed to exploit the tendency of benzo-15-crown-5 ether (B15C5) to form 2:1 stoichiometric sandwich complexes with potassium ions.76 One example reported by Taylor and co-workers features a 1,3-bis(iodotriazole)benzene-based XB ion-pair receptor 9 functionalised with B15C5 groups (Fig. 8(c)). Pre-complexation of 9 to K+ or Rb+ facilitated the formation of an intramolecular sandwich complex which led to significant enhancements of up to 700-fold in the apparent halide anion association constants of the receptor, whereas Na+-complexation resulted in only a 15-fold enhancement arising from electrostatic effects.77 Recently, Docker and co-workers reported a chalcogen bonding (ChB)98 heteroditopic receptor 10 based on a modified receptor design, which consists of an acyclic 3,5-bis-tellurotriazole nitro-benzene scaffold with B15C5 units directly appended to the tellurium-incorporated triazoles (Fig. 8(d)). 10 showed marked selectivity for potassium chloride over other alkali metal chloride salts, with crystallographic studies confirming the formation of an intramolecular co-facial bis-B15C5 K+ sandwich complex. In addition, 10 was shown to solubilise KCl in CDCl3 through a series of solid–liquid and liquid–liquid extraction studies, and preliminary U-tube membrane transport experiments further indicated its capability to selectively transport KCl across a membrane.60
Tse and co-workers prepared a series of heteroditopic macrocycles comprising a phenanthroline-based cation binding site opposite a bidentate anion binding motif based on either halogen bond (11XB), chalcogen bond (11ChB) or hydrogen bond (11HB) donor motifs (Fig. 9(a)).571H NMR titration studies in CDCl3/CD3CN solvent mixtures indicated strong cooperative binding of lithium halide ion-pairs by 11XB and 11ChB, which exhibited enhanced binding affinities relative to 11HB. Importantly, the ion-pair binding affinities and preferences of the macrocycles could be tuned by varying the nature of the anion binding interaction. 11XB showed overall strongest binding affinity for all LiX ion-pairs (X = Cl, Br, I) and is notably able to stabilise the ‘hard’ LiCl ion-pair. In contrast, 11ChB was unable to bind LiCl and instead demonstrates a marked preference for the ‘soft’ LiI ion-pair.
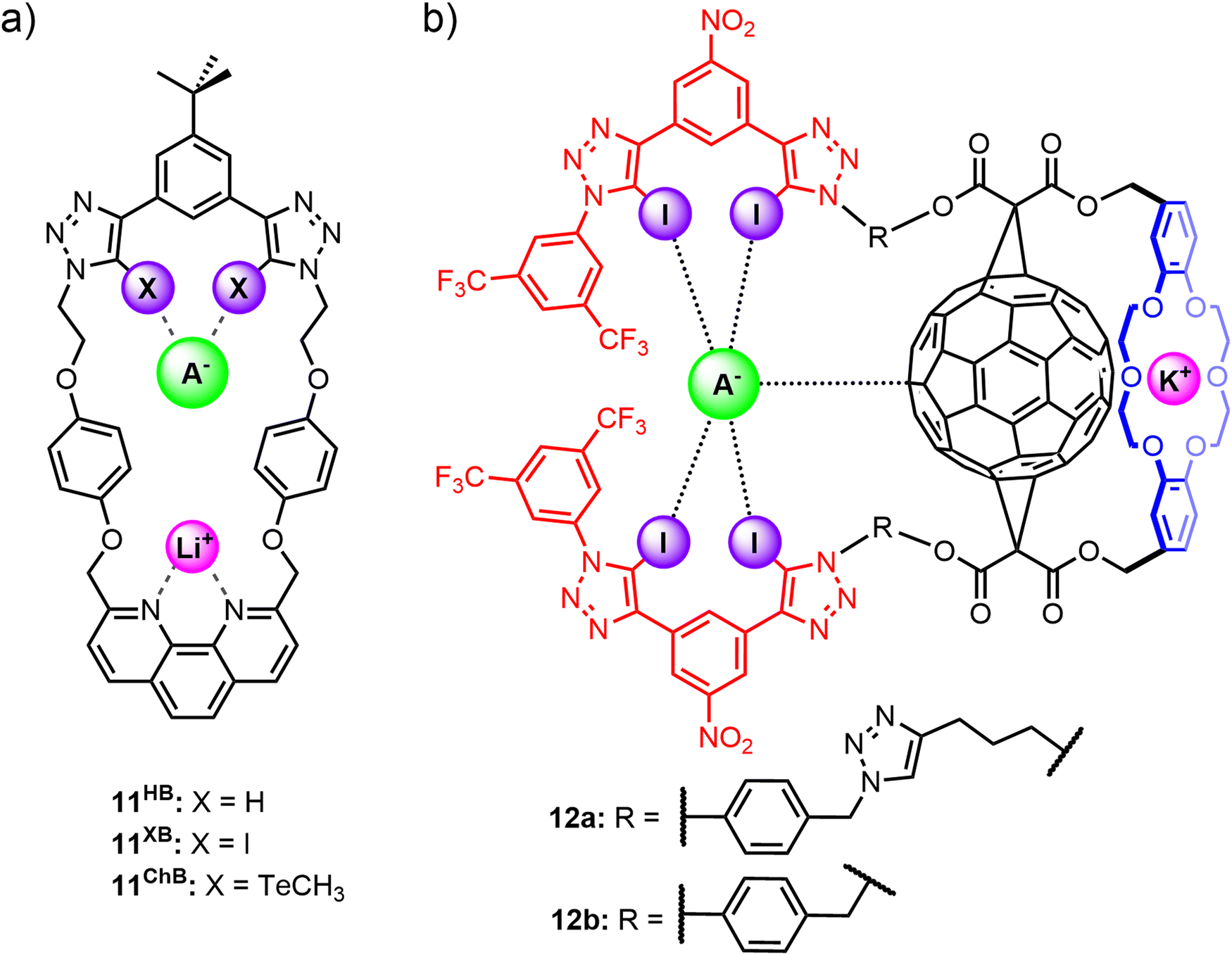 | ||
| Fig. 9 Non-interlocked heteroditopic incorporating XB, ChB and HB anion binding motifs, including (a) Tse's phenanthroline-functionalised macrocycles 11, and (b) Bak's fullerene-containing receptors 12a–b. | ||
Another example of integrating XB⋯anion interactions into a macrocyclic scaffold for alkali metal halide ion-pair recognition was reported by Bak and co-workers,105 who synthesised fullerene-containing heteroditopic receptors 12a–b utilising iodotriazole XB donors as the anion binding motif and dibenzo-18-crown-6 ether as the cation binding group, wherein importantly the cation and anion binding sites are spatially separated by a C60 unit (Fig. 9(b) and (c)). The neutral receptors 12a–b displayed enhanced XB-mediated anion binding relative to a non-fullerene analogue in 3:1 CD3CN/CDCl3, attributed to a combination of fullerene-mediated pre-organisation, solvent shielding and the formation of π–hole interactions with the polarisable C60 surface. Pre-complexation of a potassium cation to the crown ether unit significantly enhanced the halide binding affinities of 12a–b through positive electrostatic cooperativity associated with the increased polarisation of the C60 surface by the co-bound cation. The important contribution from C60 polarisation was reflected in the marked binding preference of the potassium bound receptor complex 12·K+ for the softer iodide ion (Ka(I−) = 36000 M−1 (12a), 19800 M−1 (12b)) over bromide (Ka(Br−) = 25600 M−1 (12a), 8300 M−1 (12b)) and chloride (Ka(Cl−) = 15100 M−1 (12a), 9300 M−1 (12b)). Interestingly, detailed analysis of the 1H NMR spectral data suggests that a significant proportion (40%) of the chloride guest is directly coordinated to the potassium cation in a contact ion-pair binding fashion, and does not benefit from the polarisation-enhanced XB interactions, accounting for the halide's weaker binding to the receptor. These studies thereby highlight the potential of integrating XB interactions into heteroditopic receptor design to elicit nuanced selectivity effects in ion-pair binding.
MIM heteroditopic receptors utilising σ-hole interactions
To the best of our knowledge, the first instance of a heteroditopic [2]rotaxane-based ion-pair receptor 13 employing XB donor groups for anion binding was reported by our group in 2022.106 The rotaxane design consists of a bis(iodotriazole)-functionalised macrocycle as well as an iodotriazole-functionalised axle component (Fig. 10(a)), which was prepared via a CuAAC active metal template reaction. Quantitative 1H NMR studies showed that 13 was capable of binding lithium bromide and iodide ion-pairs in CDCl3/CD3CN mixtures, an impressive feat considering the high lattice enthalpies of lithium halide salts. Crystallographic and DFT modelling studies indicated that the interlocked nature of 13 was pivotal in creating the requisite cation and anion binding sites. Cation binding occurs via the 2,6-dialkoxypyridyl motif of the macrocycle and the triazole N-donors of the axle, which forms a binding cavity complementary in size to the small Li+ cation. Li+ complexation within this cavity pre-organises the iodotriazole XB motifs of the macrocycle and axle to participate in convergent anion binding, giving rise to a positive allosteric effect that contributes to favourable cooperativity in ion-pair binding. Importantly, the non-interlocked axle and macrocycle components were unable to bind lithium halide ion-pairs. The selectivity of the rotaxanes for binding lithium halide ion-pairs is particularly interesting given the widespread adoption of lithium-based battery technologies,107 which has in turn driven efforts to develop efficient methods for the recovery and remediation of lithium salts.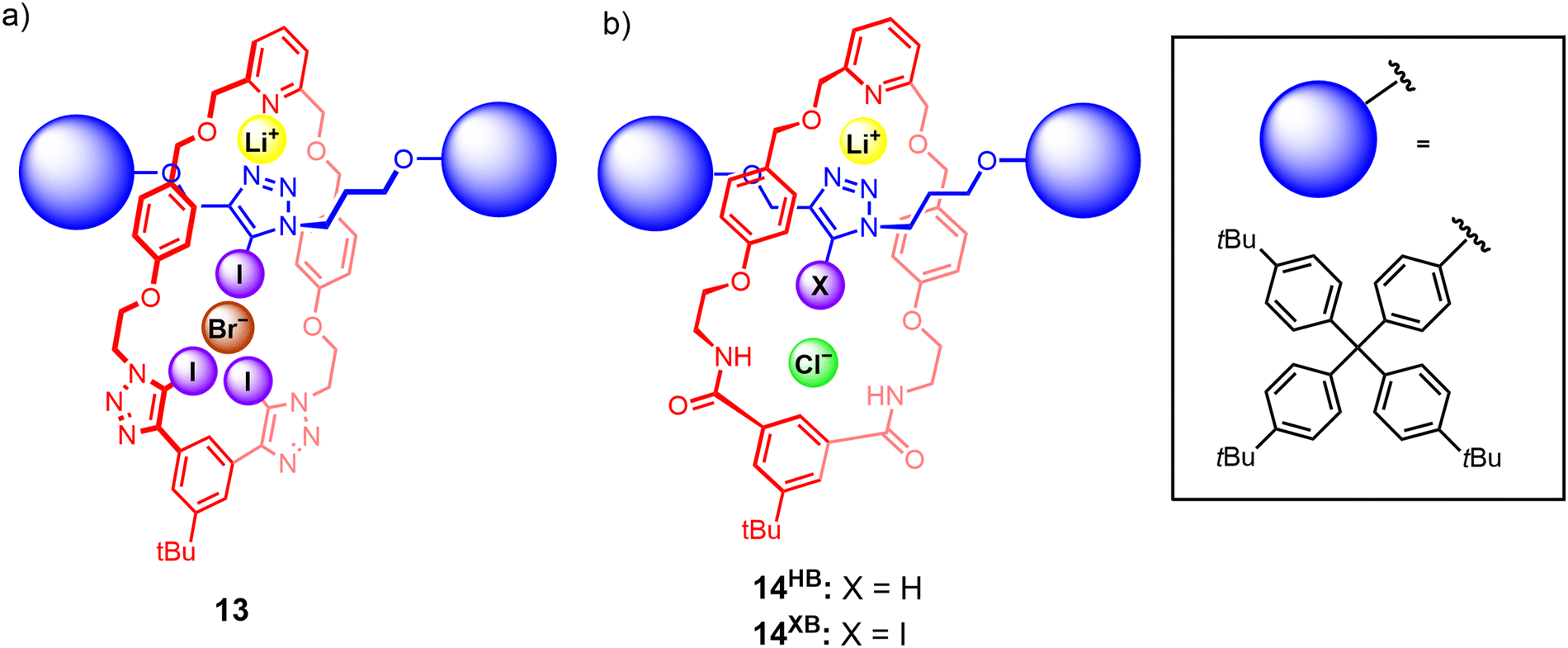 | ||
| Fig. 10 Beer's heteroditopic XB [2]rotaxanes capable of cooperatively binding LiX ion-pairs, including: (a) rotaxane 13 which preferentially binds LiBr and LiI ion-pairs; (b) rotaxanes 14HB and 14XB which are capable of stabilising LiCl ion-pairs. | ||
Subsequently, related rotaxanes 14HB and 14XB employing a similar design concept, with an isophthalamide-based macrocycle and an axle containing a prototriazole (14HB) or iodotriazole (14XB) motif, displayed remarkably selective binding of lithium chloride over other alkali metal halides (Fig. 10(b)). Interestingly, 1H NMR, crystallographic and DFT computational studies revealed the free rotaxanes adopted a co-conformation which allows the axle triazole nitrogen atoms to participate in intramolecular hydrogen bonding interactions with the isophthalamide motifs. Binding of Li+ induces a re-orientation of the triazole to create pre-organised cation and anion binding cavities, thereby exploiting the co-conformational dynamism of the MIM architecture to achieve a positive cooperative allosteric enhancement of anion binding upon Li+ pre-complexation.
Integrating XB donor motifs into pre-organised catenane-based host architectures proved to be a promising strategy for modulating the binding affinities and selectivity profiles of ion-pair binding (Fig. 11). A series of neutral all-XB homo[2]catenanes 15DEG and 15TEG were prepared via an elegant template-directed synthetic strategy, in which a sodium cation directs the formation of a pseudo-[2]rotaxane complex between an oligo(ethylene glycol)-functionalised XB macrocycle and a bis-azide. The acyclic bis-azide precursor subsequently underwent a CuAAC-mediated macrocyclisation reaction with a bis-iodoalkyne to form a second interlocked macrocycle containing a bidentate XB donor motif. The mechanical bond formation step thereby simultaneously generated both cation and anion recognition sites (Fig. 11(a)).
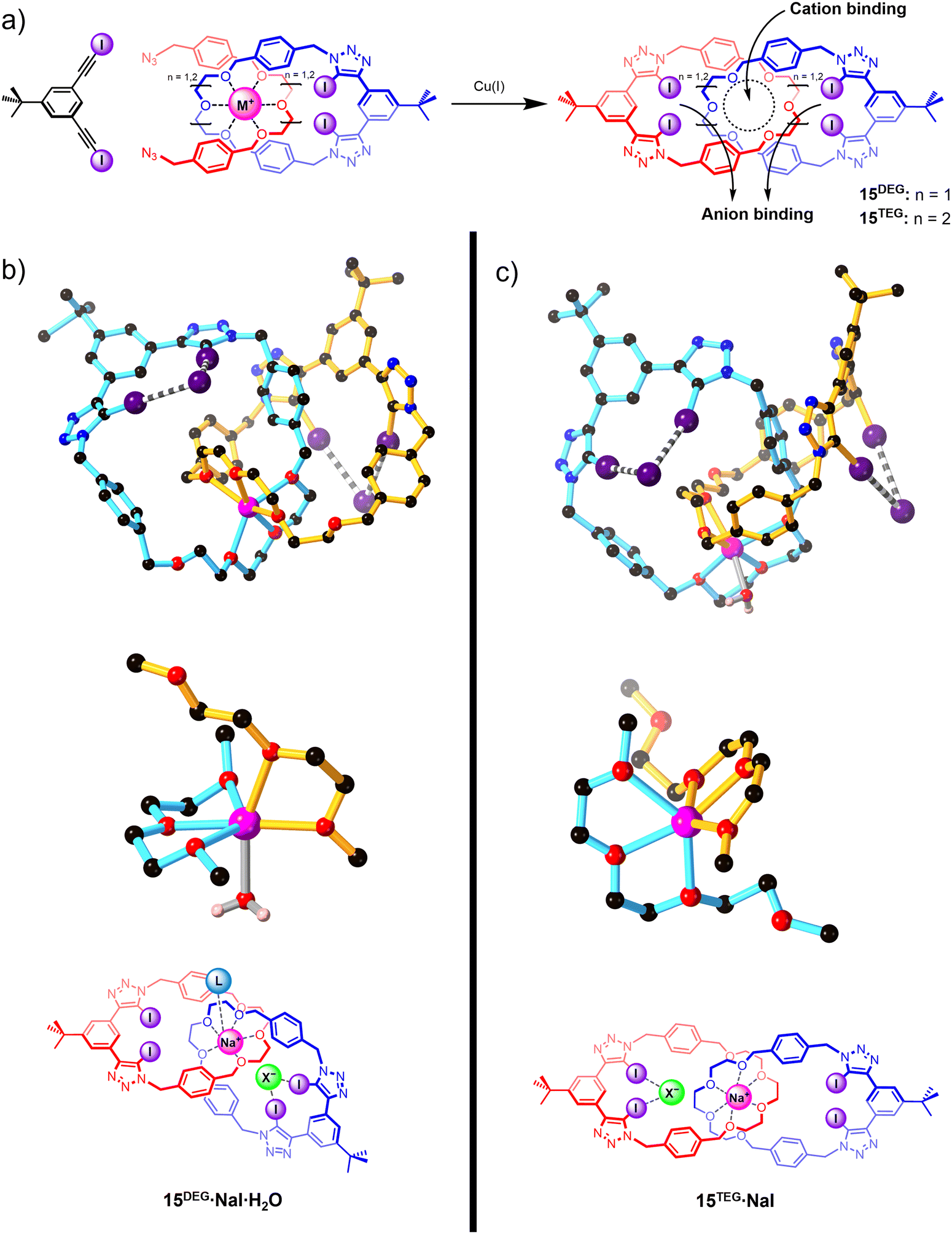 | ||
| Fig. 11 (a) Sodium cation template-directed synthesis of heteroditopic all-XB [2]catenanes 15 for alkali metal halide ion-pair binding; solid-state structures of (b) 15DEG·NaI·H2O and (c) 15TEG·NaI, showing a single [2]catenane unit bound to a sodium cation and two iodide anions (top) and a truncated representation of the ethylene glycol region (middle) highlighting the coordination environment around the sodium ion. Atom colours are as follows: black (carbon), blue (nitrogen), red (oxygen), purple (iodine), light pink (hydrogen), bright pink (sodium). The chemical structures of 15DEG and 15TEG are shown below for reference. | ||
The resulting homo[2]catenanes exhibited moderate affinity for sodium and potassium cations, with 1:1 host–guest stoichiometric binding constants (Ka) in the range 700–1800 M−1 in 1:1 CDCl3/CD3CN solvent mixtures. Notably, the smaller di(ethylene glycol)-based catenane 15DEG preferentially binds sodium over potassium and vice versa for the larger tri(ethylene glycol)-based catenane 15TEG, reflecting the size complementarity between the interlocked cation binding site and the cation guests. No metal cation binding was observed for the non-interlocked macrocycles, attesting to the importance of the mechanical bond effect (MBE) in switching on the cation affinity of the host. The modest halide affinities of the free catenanes (Ka = 100–200 M−1) were found to be significantly enhanced by pre-complexation of a sodium or potassium cation, reflecting the positive electrostatic cooperativity in ion-pair binding. Interestingly, a detailed analysis of the 1H NMR titration isotherms showed that depending on the identity of the catenane and the pre-complexed metal ion, the halides were bound in either a 1:1 or 1:2 host–guest stoichiometry. XRD crystallographic analysis revealed that, in addition to the anticipated XB-mediated anion binding, the smaller [2]catenane 15DEG can also adopt a direct contact ion-pair binding mode in which an anion guest coordinates to the unsaturated Na+ centre (Fig. 11(b)), giving rise to an overall 1:1:2 host–cation–anion stoichiometry. In contrast, the larger [2]catenane 15TEG exhibits a 1:1:1 host–cation–anion binding stoichiometry as its coordinatively saturated Na+ cation guest precludes direct contact ion-pair binding (Fig. 11(c)).108
While the homo[2]catenane systems highlighted the potential of exploiting the highly pre-organised binding cavities of catenanes to elicit ion-pair binding, the relatively moderate individual cation and anion affinities of 15DEG and 15TEG precluded strong ion-pair binding. Building on this work, a heteroditopic [2]catenane ion-pair host system 16 containing a more potent bis-di(ethylene glycol) cation binding site was synthesised (Fig. 12), which demonstrated a dramatic increase in affinity for sodium and potassium cations compared to homo[2]catenane systems, as well as marked sodium over potassium selectivity arising from the size complementarity between the di(ethylene glycol)-based cation binding site and the smaller sodium cation. Similar to the homo[2]catenanes, ion-pair titrations conducted on 16 in 1:1 or 1:3 CDCl3/CD3CN solvent mixtures showed that its anion binding affinity could be ‘switched on’ by pre-complexation with a metal cation. Notably, 16 displayed significantly stronger binding of alkali metal halide salts compared to the homo[2]catenanes, indicating that the modified host design was effective in enhancing the ion-pair affinity of the interlocked receptor. In addition, the binding preference for sodium halide over potassium halide ion-pairs was maintained, with 16 displaying 3 to 5-fold stronger binding to NaX ion-pairs (X = Br, I) relative to the corresponding KX salts. This binding preference was impressively translated to the solid–liquid extraction properties of the catenane, with competitive SLE experiments indicating preferential extraction of sodium halide salts from a solid mixture of NaX/KX salts (X = Br, I).109
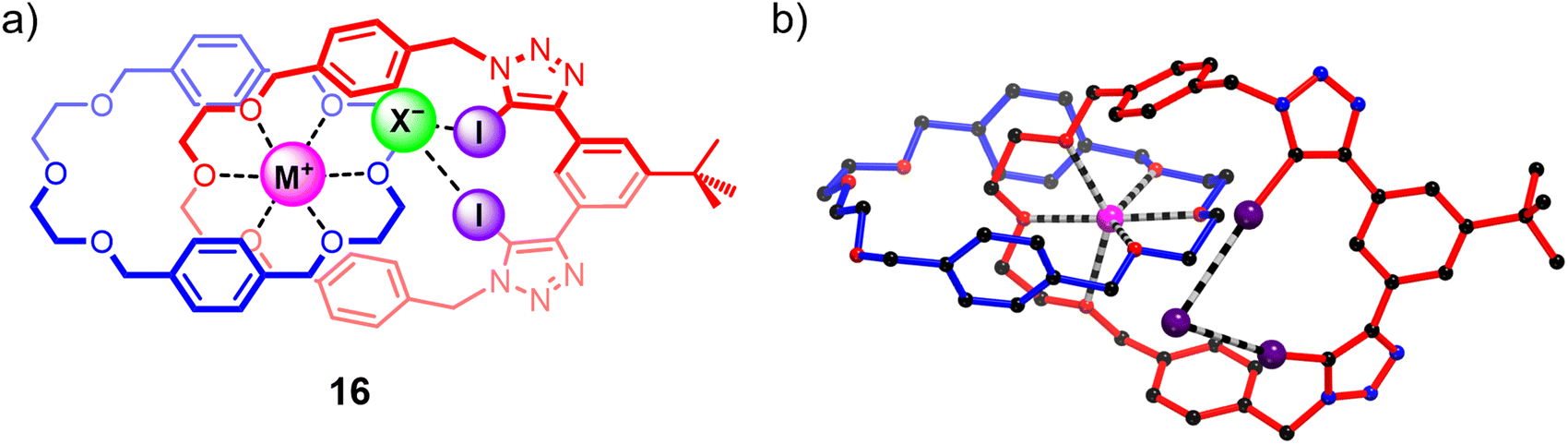 | ||
| Fig. 12 (a) A heteroditopic XB [2]catenane 16 showing enhanced alkali metal halide ion-pair binding and extraction properties. (b) Crystal structure of 16 bound to an NaI ion-pair in a host-separated binding mode. | ||
To the best of our knowledge, comparative studies of ‘mechanical bond isomers’ for ion-pair binding and recognition remain unprecedented to date. This is somewhat surprising considering the different co-conformational behaviours of rotaxanes and catenanes and the potential effects on their recognition properties. As such, encouraged by the promising ion-pair binding properties of the catenane-based heteroditopic receptors, attention was turned to investigating their [2]rotaxane analogues (Fig. 13(a)). To this end, a series of heteroditopic [2]rotaxanes 17a–d were prepared via a modified sodium cation template-directed methodology, in which a stoppering-type reaction was performed on the intermediate pseudo[2]rotaxane assembly using sterically bulky iodoalkyne-functionalised terphenyl groups. Notably, 1H NMR binding studies revealed that metal cation binding to the rotaxanes induces a translocation of the macrocycle component towards the central ethylene glycol stations to facilitate convergent metal coordination by the polyether binding motifs of the macrocycle and axle. The need for a large-amplitude co-conformational change led to reduced cation binding affinities of the [2]rotaxanes relative to the analogous homo[2]catenanes, which in turn gave rise to complex parallel and competing equilibria in the ion-pair binding studies that complicated the quantitative determination of ion-pair binding affinities. Importantly, comparing the apparent alkali metal cation iodide ion-pair binding constants of the [2]rotaxanes and the [2]catenane analogues revealed that the rotaxanes exhibited a higher degree of positive cooperativity in ion-pair binding despite weaker cation binding (Fig. 13(b)), suggesting the advantages of incorporating mechanical bond co-conformational flexibility into the design of future heteroditopic interlocked receptors.110
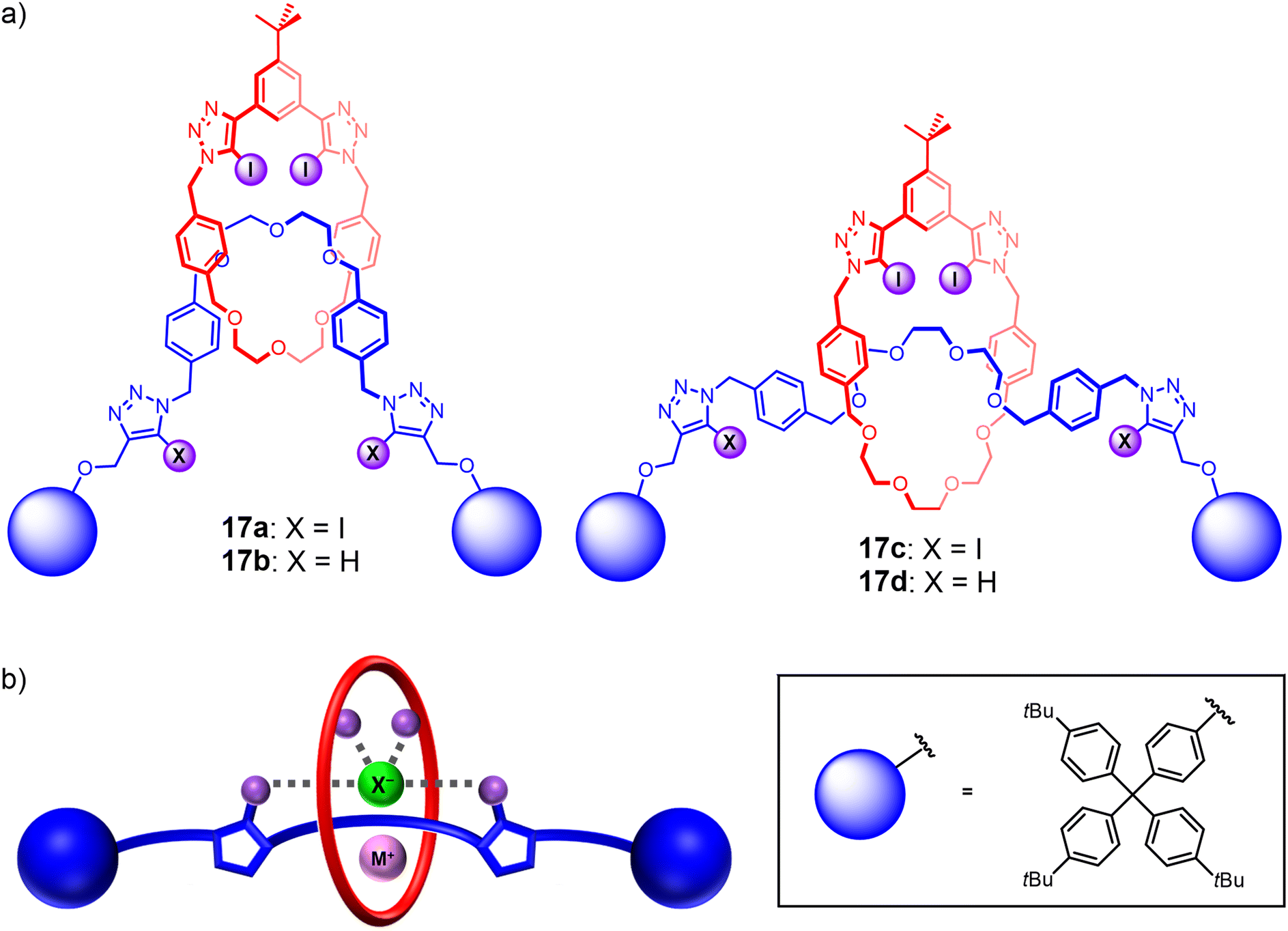 | ||
| Fig. 13 (a) Heteroditopic HB and XB rotaxanes 17a–d demonstrating cooperative binding of alkali metal halide ion-pairs; (b) proposed ion-pair binding mode of heteroditopic rotaxanes. | ||
Conclusions
In the pursuit of novel heteroditopic receptors capable of improved ion-pair binding affinity and selectivity, the use of mechanically interlocked host topologies is emerging as a promising avenue of research. Although efforts to incorporate the mechanical bond into heteroditopic host design have thus far remained limited, the examples discussed in this review importantly highlight significant binding affinity enhancements and positive cooperativity in ion-pair recognition by MIM-based heteroditopic receptors relative to non-interlocked acyclic and macrocyclic analogues. Crucially, in many cases, MIM-based heteroditopic receptors leverage not only favourable proximal cation–anion bound electrostatic effects but also their inherent co-conformational dynamism to achieve allosteric cooperativity, wherein the binding of one guest ion induces a co-conformational rearrangement of the mechanically interlocked components, including macrocycle translocation, pirouetting or inter-ring circum-rotation, to create a pre-organised binding cavity suited for binding to the second guest counter-ion of the ion-pair. Impressively, chemically similar alkali metal cations or halide anions can often be distinguished by varying the nature of the cation/anion binding motifs and the size of the interlocked cavity, which serves as a powerful means to achieve highly selective ion-pair binding. Notably, recent advancements in the incorporation of halogen bonding (XB) anion recognition motifs into MIM receptor scaffolds have enabled further modulation of ion-pair binding affinity and selectivity profiles.Given the relatively small number of heteroditopic MIM receptors reported to date, future work in this area is expected to initially focus on laying the groundwork for this expanding field. From a synthetic perspective, this would entail exploiting and combining emerging classes of non-covalent interactions, such as chalcogen bonding and anion–π interactions in innovative host designs. Equally important is the development of robust analytical methods to quantify the strength and cooperativity of ion-pair binding, which would enable systematic comparisons to be made between various receptor systems. Combining these synthetic and analytical tools to evaluate the cation, anion and electrostatic/allosteric cooperative ion-pair binding properties would elucidate guiding principles to achieve selective ion-pair recognition in future strategic MIM heteroditopic host design.
Further progress in the field would encourage the construction of functional MIM-based ion-pair receptors for a diverse range of applications. For example, MIM-based heteroditopic architectures are particularly suited for use as ion-pair sensors, as their high binding affinity and selectivity for target guests allows them to function at low concentrations. Initially, sensors can be prepared in a relatively facile manner via the integration of fluorophoric or redox-active reporter groups into established heteroditopic MIM receptor scaffolds. Additionally, the inherent co-conformational dynamism of MIMs is a powerful tool for the development of novel sensing mechanisms for ion-pairs, as has been demonstrated in a series of reports featuring chromophore-functionalised rotaxane shuttles which utilise anion recognition induced macrocycle translocation to elicit a selective optical response to specific anionic guest species.48,111–115 There is also potential for utilising MIM-based heteroditopic receptors in selective ion-pair transport or extraction. Finally, the scope for guest binding may further expand from simple inorganic ion-pairs to the recognition of complex guest species such as biological zwitterions including amino acids, where chiral heteroditopic MIM structural host frameworks can be employed.116–118
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. M. T. acknowledges the Clarendon Fund and the Oxford Australia Scholarships Fund for a postgraduate research scholarship.Notes and references
- D. Banerjee, D. Kim, M. J. Schweiger, A. A. Kruger and P. K. Thallapally, Chem. Soc. Rev., 2016, 45, 2724–2739 RSC.
- M. D. Rao, K. K. Singh, C. A. Morrison and J. B. Love, RSC Adv., 2020, 10, 4300–4309 RSC.
- M. P. Anderson, R. J. Gregory, S. Thompson, D. W. Souza, S. Paul, R. C. Mulligan, A. E. Smith and M. J. Welsh, Science, 1991, 253, 202 CrossRef CAS PubMed.
- F. Delange, Thyroid, 1994, 4, 107–128 CrossRef CAS PubMed.
- J. L. Way, Annu. Rev. Pharmacol. Toxicol., 1984, 24, 451–481 CrossRef CAS PubMed.
- V. H. Smith and D. W. Schindler, Trends. Ecol. Evol., 2009, 24, 201–207 CrossRef PubMed.
- J. H. P. Watson and D. C. Ellwood, Nucl. Eng. Des., 2003, 226, 375–385 CrossRef CAS.
- P. K. Dasgupta, J. V. Dyke, A. B. Kirk and W. A. Jackson, Environ. Sci. Technol., 2006, 40, 6608–6614 CrossRef CAS PubMed.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed.
- F. P. Schmidtchen, Angew. Chem., Int. Ed. Engl., 1977, 16, 720–721 CrossRef.
- E. Graf and J. M. Lehn, J. Am. Chem. Soc., 1976, 98, 6403–6405 CrossRef CAS.
- J. M. Lehn, E. Sonveaux and A. K. Willard, J. Am. Chem. Soc., 1978, 100, 4914–4916 CrossRef CAS.
- G. Müller, J. Riede and F. P. Schmidtchen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1516–1518 CrossRef.
- K. Chellappan, N. J. Singh, I.-C. Hwang, J. W. Lee and K. S. Kim, Angew. Chem., Int. Ed., 2005, 44, 2899–2903 CrossRef CAS PubMed.
- A. Kumar and P. S. Pandey, Org. Lett., 2008, 10, 165–168 CrossRef CAS PubMed.
- J. T. Davis, P. A. Gale and R. Quesada, Chem. Soc. Rev., 2020, 49, 6056–6086 RSC.
- N. Akhtar, O. Biswas and D. Manna, Chem. Commun., 2020, 56, 14137–14153 RSC.
- K. M. Bąk, K. Porfyrakis, J. J. Davis and P. D. Beer, Mater. Chem. Front., 2020, 4, 1052–1073 RSC.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed.
- J. E. M. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2017, 53, 298–312 RSC.
- W.-X. Gao, H.-J. Feng, B.-B. Guo, Y. Lu and G.-X. Jin, Chem. Rev., 2020, 120, 6288–6325 CrossRef CAS PubMed.
- H.-N. Zhang, Y.-J. Lin and G.-X. Jin, J. Am. Chem. Soc., 2021, 143, 1119–1125 CrossRef CAS PubMed.
- J. T. Wilmore and P. D. Beer, Adv. Mater., 2024, 36, 2309098 CrossRef CAS PubMed.
- G. Baggi and S. J. Loeb, Chem. – Eur. J., 2017, 23, 14163–14166 CrossRef CAS PubMed.
- M. Nandi, S. Bej, T. K. Ghosh and P. Ghosh, Chem. Commun., 2019, 55, 3085–3088 RSC.
- S.-M. Chan, F.-K. Tang, C.-S. Kwan, C.-Y. Lam, S. C. K. Hau and K. C.-F. Leung, Mater. Chem. Front., 2019, 3, 2388–2396 RSC.
- T. Shukla, A. K. Dwivedi, R. Arumugaperumal, C.-M. Lin, S.-Y. Chen and H.-C. Lin, Dyes Pigm., 2016, 131, 49–59 CrossRef CAS.
- M. Denis, J. Pancholi, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5310–5314 CrossRef CAS PubMed.
- A. Caballero, F. Zapata and P. D. Beer, Coord. Chem. Rev., 2013, 257, 2434–2455 CrossRef CAS.
- R. J. Goodwin, A. Docker, H. I. MacDermott-Opeskin, H. M. Aitken, M. L. O'Mara, P. D. Beer and N. G. White, Chem. – Eur. J., 2022, 28, e202200389 CrossRef CAS PubMed.
- J. M. Mercurio, F. Tyrrell, J. Cookson and P. D. Beer, Chem. Commun., 2013, 49, 10793–10795 RSC.
- M. J. Langton, O. A. Blackburn, T. Lang, S. Faulkner and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11463–11466 CrossRef CAS PubMed.
- N. G. White and P. D. Beer, Org. Biomol. Chem., 2013, 11, 1326–1333 RSC.
- Y. Cheong Tse, R. Hein, E. J. Mitchell, Z. Zhang and P. D. Beer, Chem. – Eur. J., 2021, 27, 14550–14559 CrossRef CAS PubMed.
- J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- F. Bitsch, C. O. Dietrich-Buchecker, A. K. Khemiss, J. P. Sauvage and A. Van Dorsselaer, J. Am. Chem. Soc., 1991, 113, 4023–4025 CrossRef CAS.
- C. O. Dietrich-Buchecker, C. Hemmert, A. K. Khemiss and J. P. Sauvage, J. Am. Chem. Soc., 1990, 112, 8002–8008 CrossRef CAS.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1989, 28, 189–192 CrossRef.
- J. Guilhem, C. Pascard, J. P. Sauvage and J. Weiss, J. Am. Chem. Soc., 1988, 110, 8711–8713 CrossRef CAS.
- J. P. Sauvage and J. Weiss, J. Am. Chem. Soc., 1985, 107, 6108–6110 CrossRef CAS PubMed.
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. M. Kern, J. Am. Chem. Soc., 1984, 106, 3043–3045 CrossRef CAS.
- M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, J. Am. Chem. Soc., 2019, 141, 879–889 CrossRef CAS PubMed.
- A. Docker, Y. C. Tse, H. M. Tay, A. J. Taylor, Z. Zhang and P. D. Beer, Angew. Chem., Int. Ed., 2022, 61, e202214523 CrossRef CAS PubMed.
- H. M. Tay, T. G. Johnson, A. Docker, M. J. Langton and P. D. Beer, Angew. Chem., Int. Ed., 2023, 62, e202312745 CrossRef PubMed.
- H. M. Tay, A. Docker, A. Taylor and P. D. Beer, Chem. – Eur. J., 2024, 30, e202400952 CrossRef CAS PubMed.
- K. M. Bąk, B. Trzaskowski and M. J. Chmielewski, Chem. Sci., 2024, 15, 1796–1809 RSC.
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC.
- A. J. McConnell, A. Docker and P. D. Beer, ChemPlusChem, 2020, 85, 1824–1841 CrossRef CAS PubMed.
- P. D. Beer, P. K. Hopkins and J. D. McKinney, Chem. Commun., 1999, 1253–1254 RSC.
- M. Zakrzewski, D. Załubiniak and P. Piątek, Dalton Trans., 2018, 47, 323–330 RSC.
- Q. He, G. M. Peters, V. M. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2017, 56, 13396–13400 CrossRef CAS PubMed.
- S. G. Galbraith, P. G. Plieger and P. A. Tasker, Chem. Commun., 2002, 2662–2663 RSC.
- T. Bunchuay, A. Docker, U. Eiamprasert, P. Surawatanawong, A. Brown and P. D. Beer, Angew. Chem., Int. Ed., 2020, 59, 12007–12012 CrossRef CAS PubMed.
- Y. C. Tse, A. Docker, Z. Zhang and P. D. Beer, Chem. Commun., 2021, 57, 4950–4953 RSC.
- A. Docker, J. G. Stevens and P. D. Beer, Chem. – Eur. J., 2021, 27, 14600–14604 CrossRef CAS PubMed.
- A. Docker, T. Bunchuay, M. Ahrens, A. J. Martinez-Martinez and P. D. Beer, Chem. – Eur. J., 2021, 27, 7837–7841 CrossRef CAS PubMed.
- A. Docker, I. Marques, H. Kuhn, Z. Zhang, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2022, 144, 14778–14789 CrossRef CAS PubMed.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed.
- Q. He, Z. Zhang, J. T. Brewster, V. M. Lynch, S. K. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 9779–9782 CrossRef CAS PubMed.
- A. V. Koulov, J. M. Mahoney and B. D. Smith, Org. Biomol. Chem., 2003, 1, 27–29 RSC.
- J. M. Mahoney, G. U. Nawaratna, A. M. Beatty, P. J. Duggan and B. D. Smith, Inorg. Chem., 2004, 43, 5902–5907 CrossRef CAS PubMed.
- A. Galan, D. Andreu, A. M. Echavarren, P. Prados and J. De Mendoza, J. Am. Chem. Soc., 1992, 114, 1511–1512 CrossRef CAS.
- W. Walczak, M. Zakrzewski, G. Cichowicz and P. Piątek, Org. Biomol. Chem., 2020, 18, 694–699 RSC.
- R. C. Knighton and P. D. Beer, Chem. Commun., 2014, 50, 1540–1542 RSC.
- A. Brown, K. M. Mennie, O. Mason, N. G. White and P. D. Beer, Dalton Trans., 2017, 46, 13376–13385 RSC.
- M. J. Deetz, R. Shukla and B. D. Smith, Tetrahedron, 2002, 58, 799–805 CrossRef CAS.
- J. R. Romero, G. Aragay and P. Ballester, Chem. Sci., 2017, 8, 491–498 RSC.
- M. Denis, L. Qin, P. Turner, K. A. Jolliffe and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5315–5319 CrossRef CAS PubMed.
- D.-H. Li and B. D. Smith, J. Org. Chem., 2019, 84, 2808–2816 CrossRef CAS PubMed.
- A. J. McConnell and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 5052–5061 CrossRef CAS PubMed.
- J. L. Sessler, S. K. Kim, D. E. Gross, C.-H. Lee, J. S. Kim and V. M. Lynch, J. Am. Chem. Soc., 2008, 130, 13162–13166 CrossRef CAS PubMed.
- J. Scheerder, J. P. M. van Duynhoven, J. F. J. Engbersen and D. N. Reinhoudt, Angew. Chem., Int. Ed. Engl., 1996, 35, 1090–1093 CrossRef.
- A. J. Evans and P. D. Beer, Dalton Trans., 2003, 4451–4456 RSC.
- A. J. Taylor, A. Docker and P. D. Beer, Chem. – Asian J., 2023, 18, e202201170 CrossRef CAS PubMed.
- M. J. Deetz, M. Shang and B. D. Smith, J. Am. Chem. Soc., 2000, 122, 6201–6207 CrossRef CAS.
- J. M. Mahoney, A. M. Beatty and B. D. Smith, J. Am. Chem. Soc., 2001, 123, 5847–5848 CrossRef CAS PubMed.
- Y. Yeon, S. Leem, C. Wagen, V. M. Lynch, S. K. Kim and J. L. Sessler, Org. Lett., 2016, 18, 4396–4399 CrossRef CAS PubMed.
- B. Akhuli and P. Ghosh, Chem. Commun., 2015, 51, 16514–16517 RSC.
- D. Jagleniec, S. Siennicka, Ł. Dobrzycki, M. Karbarz and J. Romański, Inorg. Chem., 2018, 57, 12941–12952 CrossRef CAS PubMed.
- P. R. A. Webber and P. D. Beer, Dalton Trans., 2003, 2249–2252 RSC.
- Z.-H. Sun, F.-F. Pan, Triyanti, M. Albrecht and G. Raabe, Eur. J. Org. Chem., 2013, 7922–7932 CrossRef CAS.
- J. Romański and P. Piątek, J. Org. Chem., 2013, 78, 4341–4347 CrossRef PubMed.
- J. H. Lee, J. H. Lee, Y. R. Choi, P. Kang, M.-G. Choi and K.-S. Jeong, J. Org. Chem., 2014, 79, 6403–6409 CrossRef CAS PubMed.
- D.-y Duan, L. L. H. Liu, N. Bozeat, Z. M. Huang, S. Y. Xiang, G.-l Wang, L. Ye and J. R. Hume, Acta Pharmacol. Sin., 2005, 26, 265–278 CrossRef CAS PubMed.
- L. Puljak and G. Kilic, Biochim. Biophys. Acta, Mol. Basis Dis., 2006, 1762, 404–413 CrossRef CAS PubMed.
- N. Carrasco, Biochim. Biophys. Acta, Rev. Biomembr., 1993, 1154, 65–82 CrossRef CAS PubMed.
- X. Zheng, Z. Zhu, X. Lin, Y. Zhang, Y. He, H. Cao and Z. Sun, Engineering, 2018, 4, 361–370 CrossRef CAS.
- M. Kamenica, R. R. Kothur, A. Willows, B. A. Patel and P. J. Cragg, Sensors, 2017, 17, 2430 CrossRef PubMed.
- R. C. Knighton and P. D. Beer, Org. Chem. Front., 2021, 8, 2468–2472 RSC.
- S. K. Kim and J. L. Sessler, Acc. Chem. Res., 2014, 47, 2525–2536 CrossRef CAS PubMed.
- V. Valderrey, E. C. Escudero-Adán and P. Ballester, J. Am. Chem. Soc., 2012, 134, 10733–10736 CrossRef CAS PubMed.
- A. Arun, A. Docker, H. Min Tay and P. D. Beer, Chem. – Eur. J., 2023, 29, e202301446 CrossRef CAS PubMed.
- Y.-J. Lee, T.-H. Ho, C.-C. Lai and S.-H. Chiu, Org. Biomol. Chem., 2016, 14, 1153–1160 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- A. Docker and P. D. Beer, Halogen Bonding in Solution, 2021, pp. 83–120 Search PubMed.
- T. Bunchuay, A. Docker, A. J. Martinez-Martinez and P. D. Beer, Angew. Chem., Int. Ed., 2019, 58, 13823–13827 CrossRef CAS PubMed.
- G. Turner, A. Docker and P. D. Beer, Dalton Trans., 2021, 50, 12800–12805 RSC.
- A. Mele, P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2005, 127, 14972–14973 CrossRef CAS PubMed.
- R. Tepper, B. Schulze, P. Bellstedt, J. Heidler, H. Görls, M. Jäger and U. S. Schubert, Chem. Commun., 2017, 53, 2260–2263 RSC.
- K. M. Bąk, I. Marques, H. Kuhn, K. E. Christensen, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2023, 145, 27367–27379 CrossRef PubMed.
- V. K. Munasinghe, J. Pancholi, D. Manawadu, Z. Zhang and P. D. Beer, Chem. – Eur. J., 2022, 28, e202201209 CrossRef CAS PubMed.
- G. Martin, L. Rentsch, M. Höck and M. Bertau, Energy Storage Mater., 2017, 6, 171–179 CrossRef.
- H. M. Tay, Y. C. Tse, A. Docker, C. Gateley, A. L. Thompson, H. Kuhn, Z. Zhang and P. D. Beer, Angew. Chem., Int. Ed., 2023, 62, e202214785 CrossRef CAS PubMed.
- H. M. Tay, A. Docker, C. Hua and P. D. Beer, Chem. Sci., 2024, 15, 13074–13081 RSC.
- H. M. Tay, A. Docker, Y. Cheong Tse and P. D. Beer, Chem. – Eur. J., 2023, 29, e202301316 CrossRef PubMed.
- J. J. Gassensmith, S. Matthys, J.-J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem. – Eur. J., 2010, 16, 2916–2921 CrossRef CAS PubMed.
- T. A. Barendt, S. W. Robinson and P. D. Beer, Chem. Sci., 2016, 7, 5171–5180 RSC.
- T. A. Barendt, A. Docker, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 11069–11076 CrossRef CAS PubMed.
- T. A. Barendt, L. Ferreira, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 9026–9037 CrossRef CAS PubMed.
- T. A. Barendt, I. Rašović, M. A. Lebedeva, G. A. Farrow, A. Auty, D. Chekulaev, I. V. Sazanovich, J. A. Weinstein, K. Porfyrakis and P. D. Beer, J. Am. Chem. Soc., 2018, 140, 1924–1936 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2018, 57, 584–588 CrossRef CAS PubMed.
- R. L. Spicer, C. C. Shearman and N. H. Evans, Chem. – Eur. J., 2023, 29, e202203502 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 12228–12239 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Professor Stephen J. Loeb on the occasion of his 70th birthday. |
| This journal is © The Royal Society of Chemistry 2024 |