
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Laccase-catalyzed tyrosine click reaction with 1-methyl-4-arylurazole: rapid labeling on protein surfaces†
Keita
Nakane
ab,
Chizu
Fujimura
a,
Shogo
Miyano
b,
Zhengyi
Liu
a,
Tatsuya
Niwa
c,
Hafumi
Nishi
def,
Tetsuya
Kadonosono
 g,
Hideki
Taguchi
c,
Shusuke
Tomoshige
b,
Minoru
Ishikawa
b and
Shinichi
Sato
*ab
g,
Hideki
Taguchi
c,
Shusuke
Tomoshige
b,
Minoru
Ishikawa
b and
Shinichi
Sato
*ab
aFrontier Research Institute for Interdisciplinary Sciences, Tohoku University, 6-3 Aramaki aza-Aoba, Aoba-ku, Sendai, Miyagi 980-8578, Japan. E-mail: shinichi.sato.e3@tohoku.ac.jp
bGraduate School of Life Sciences, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan
cCell Biology Center, Institute of Innovative Research, Tokyo Institute of Technology, 4259-S2-19, Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8501, Japan
dDepartment of Applied Information Sciences, Graduate School of Information Sciences, Tohoku University, 6-3 Aramaki Aza-Aoba, Aoba-ku, Sendai, Miyagi 980-8578, Japan
eTohoku Medical Megabank Organization, Tohoku University, 2-1 Seiryo-machi, Aoba-ku, Sendai, Miyagi 980-8573, Japan
fFaculty of Core Research, Ochanomizu University, 2-1-1 Otsuka, Bunkyo-ku, Tokyo 112-8610, Japan
gSchool of Life Science and Technology, Tokyo Institute of Technology, Yokohama, Kanagawa 226-8501, Japan
First published on 12th November 2024
Abstract
Our study demonstrates the exceptional efficiency of 1-methyl-4-arylurazole (MAUra) for tyrosine labeling, optimized with laccase under mild conditions, achieving a high efficiency (kcat/Km = 7.88 × 104 M−1 s−1) with minimal oxidative side reactions and selective labeling of highly exposed tyrosine sites on proteins.
Tyrosine residues (Tyrs) are present in 3.2% of the protein sequence and are not well exposed on the protein surface due to their hydrophobic characteristics.1 Therefore, Tyrs have desirable characteristics as targets for site-selective protein labeling techniques.2,3 Tyrs are enriched at interaction interfaces and are involved in protein–protein, protein–nucleic acid, and protein–small molecule interactions.4 Numerous researchers have explored the radical labeling reactions of Tyrs in the last 15 years. These methods include the use of radicals generated by photocatalysts,5 hemin,6 enzymes7,8 for facilitating single-electron transfer (SET) reactions to modify Tyrs as well as the use of oxidants to generate radical species.9 The primary challenge in oxidatively modifying Tyrs is the reduction for side reactions that may harm the protein structure. Although we have devised protein labeling reactions using various catalysts such as horseradish peroxidase (HRP)7 and photoredox catalysts,10 robust oxidative reaction conditions involving highly activated catalysts, oxidants such as H2O2 and ammonium peroxodisulfate, and singlet oxygen produced by excited photocatalysts can induce oxidative damage to protein structures by unintended side reactions alongside Tyr labeling.11 Therefore, for practicality, it is desirable to develop a method to achieve highly efficient Tyr labeling under milder reaction conditions.
Using enzymes that can drive oxidation using dissolved oxygen (O2) is a favorable option for reactions conducted under mild conditions. Francis et al. devised a method employing tyrosinase to selectively activate highly exposed Tyrs,12 which were subsequently captured by aniline nucleophiles. Other methods, such as labeling of the resulting quinone by cycloaddition with bicyclononyne (BCN)13 and protein–protein conjugation using cysteine residues as nucleophiles,14 have also been developed. Leveraging the unique characteristics of exceptionally exposed Tyrs within N- or C-terminal peptidic tyrosine tags, this approach facilitates the selective labeling of tyrosine tags without affecting Tyrs within the protein structure. Another recent development in tyrosinase-catalyzed protein labeling is the proximity labeling approach, in which a labeling agent with a phenolic structure is electrophilically activated to react with nucleophilic residues on the protein surface.15 Selective functionalization of Tyrs in natural protein structures, as well as Tyrs in tag sequences, represents a sought-after technique for protein functionalization.
In this study, we showed that laccase, with its ability to utilize O2 as an oxidizing agent, similar to tyrosinase, can induce radicalization of the labeling reagent and effectively modify Tyrs within native protein structures (Fig. 1). The laccase-catalyzed Tyr click reaction demonstrated exceptional efficiency compared with conventional Tyr labeling methods.
 | ||
| Fig. 1 Enzyme-catalyzed Tyr click reaction. (a) Tyrosinase-catalyzed Tyr click reaction; electrophilic activation of extremely exposed Tyrs on the tag sequence. (b) This work; Tyr click reaction on the protein surface via radical activation of the labeling reagent MAUra by laccase. | ||
The reaction conditions were designed based on the labeling reagent structures obtained in our previous studies on HRP-catalyzed Tyr labeling. The redox potential of laccase (approximately 0.78 V)16 is milder than that of HRP (approximately 1.1 V),17 and the Tyr labeling reagent can be oxidized at a potential of E1/2 360–700 mV (vs. Ag/AgCl).2 Therefore, we hypothesized that Tyrs could be labeled without activating the Tyr side (800 mV vs. Ag/AgCl) by selecting an appropriate radical precursor that could be a substrate for laccase. As Tyr labeling can proceed via an electrochemical approach that generates radical species at lower potentials than that required for directly oxidizing Tyrs on an electrode supports this hypothesis.2,18
Tyrosinase requires direct activation of Tyrs of the substrate, limiting its application to extremely exposed tyrosine tags. However, by radical activation of the low molecular weight modifier side, radical species diffusing in close proximity to the laccase active center can also label tyrosine residues in the protein structure. We previously showed that N–Me Lumi, which was developed for HRP-catalyzed Tyr labeling, can also modify Tyrs in protein structures even under laccase-catalyzed conditions.8 However, considering that there is room for improvement in the selection of suitable labeling reagents for laccase-catalyzed Tyr, we tested the Oxidant-Reduction Compound Library (targetMol Inc., 118 compounds), which is believed to be enriched with redox-active compounds, and our previously developed Tyr labeling reagent candidates (structures shown in Fig. S1 and S2, ESI†).
We screened for the compounds under the reaction conditions of 100 μM angiotensin II (1, DRVYIHPF), 1.0 equivalent of the candidate, and laccase (2.5 U mL−1, see Fig. S3 and S4 (ESI†) for enzymatic activity). After 1 h of the reaction, the candidate adducts were detected by MALDI-TOF MS. Most candidate compounds did not modify Tyr, but the urazole-type labeling reagent functioned efficiently (Fig. 2). Particularly, 1-methyl-4-phenylurazole (MAUra) and its azide derivative showed high reactivity. Because MAUra is oxidized at 570 mV (vs. Ag/AgCl)2 and produces radical species, it can be oxidized by laccase.
 | ||
| Fig. 2 Structural comparison of MAUra derivatives in Tyr labeling catalyzed by laccase. * 8.2 U mL−1 laccase was used for 7. | ||
We estimated the reaction mechanism of the reaction using MAUra. The reaction rate increased in a MAUra concentration-dependent manner, suggesting that laccase activates MAUra but not Tyr (Fig. S5a, ESI†). The catalyst constant calculated for different concentrations of MAUra is kcat/KM = 7.88 × 104 [M−1 s−1],19 which is surprisingly efficient considering that the rate constants for Cu(I)-catalyzed alkyne-azide cycloaddition (CuAAC) are 10–200 [M−1 s−1] (Fig. S5b, ESI†), indicating surprisingly efficient Tyr labeling. This reaction was inhibited by the radical scavenger, BHT, suggesting the involvement of radicals (Fig. S6, ESI†). Laccase induced MAUra degradation, suggesting that it catalyzed the radicalization reaction of MAUra (Fig. S7, ESI†). Subsequently, the effect of pH was examined. The optimum pH of laccase is 4.0–6.0,20 while MAUra has an acidic proton with a pKa of 4.721 and exists in an anionic form near neutral. Reactivity was evaluated in the presence of 10 equivalents of MAUra, and the reaction proceeded most efficiently at pH 6.0, yielding a double-modification product in which two MAUra molecules were conjugated to one Tyr. This pH dependence suggests that the anionic form of MAUra was efficiently activated by laccase (Fig. S8 and S9, ESI†).
By examining several phenol derivatives as substrates, we identified the types of phenolic structures that could be modified. The reaction proceeds even with ortho-substituted phenols (Fig. S10, ESI†). However, the reaction between phosphotyrosine and O-methyl tyrosine did not lead to modification, suggesting that the aromatic OH groups are essential labeling targets for radical species (Fig. S11, ESI†). MAUra effectively reacts with phenol derivatives like 2-nitro-para-cresol (Fig. S12, ESI†), unlike conventional reagent PTAD.22 This highlights MAUra's superior efficacy and unique reaction mechanism, allowing double modification (Fig. S13 and S14, ESI†) without side reactions to other amino acid residues. PTAD, even at higher concentrations, shows decomposition products and fails in these aspects (Fig. S15, ESI†). The usefulness of this method was demonstrated by comparing the modification of 100 μM of angiotensin II with representative reported methods. The current method, using laccase and dissolved oxygen, efficiently modifies Tyr residues with minimal side reactions, outperforming previous methods such as PTAD, electrochemical, hemin-, or HRP-catalyzed reactions, which require harsher conditions and result in undesirable by-products (see Table S1 and Fig. S15–S19, ESI†).
The selectivity of the reaction for amino acid residues was evaluated. Side reactions on lysine residues and N-terminal amino groups in the peptides did not occur. In contrast, tryptophan (Trp) residue, which is known to react with radical species, is modified under the current reaction conditions (Fig. S20 and S21, ESI†). Under conditions with both Tyr and Trp, Tyr labeling is the primary reaction. Testing on a peptide containing both residues showed that Tyr was preferentially labeled over Trp, as confirmed by HPLC and MALDI-TOF-MS (Fig. S22–S26, ESI†). In proteins, the selectivity for Tyr was even more pronounced due to Trp's minimal surface exposure.
To demonstrate the functional modification of peptides on Tyr and provide a milligram-scale supply of modified peptides, labeling reactions were performed on four Tyr-containing bioactive peptides (thymopentin, cyc(RGDyK), oxytocin, and kisspeptin-10) using compound 2 with an azide group. Efficient modifications were achieved, with single modifications preferred under low-stoichiometry conditions (Fig. S27, ESI†).
For protein-based Tyr labeling, 10 μM of protein was reacted with 3.0 equivalents of compound 2 in the presence of 8.2 U mL−1 of laccase for 1 h, followed by a 30 min reaction with 10 equivalents of DBCO-Cy3 followed by removal of the low molecular weight component. The resulting Cy3-modified proteins were digested by trypsin in gel, and peptide fragments were measured by LC-MS with a fluorescence detector to quantitatively identify the major labeled peptide fragments (Fig. S28–S32, ESI†). The labeling sites were determined by similar experiments using compound 1, and the resulting peptide fragments were identified by nanoLC-MS/MS (Fig. S34–S42, ESI†). Fig. 3 summarizes the major labeling sites of streptavidin, carbonic anhydrase II, bovine serum albumin, glucose oxidase (GOx), and trastuzumab (Fig. S43–S47, ESI†). When streptavidin and carbonic anhydrase II were used as substrates, protein recovery decreased, likely due to aggregation from increased hydrophobicity caused by the attachment of multiple 2 molecules to low molecular weight proteins (Fig. S48, ESI†). The main concern of Tyr click reactions is oxidative damage to protein structures. This method uses dissolved O2 instead of added oxidants, allowing mild reaction conditions. Free cysteine residues, sensitive to oxidative damage, were less affected by this method compared to conventional ones (Fig. S49, ESI†). Notably, this method efficiently labeled GOx, an enzyme that converts glucose to gluconolactone, with high selectivity for Y280 without impairing its activity (Fig. S31, S40, S46 and S50, ESI†).
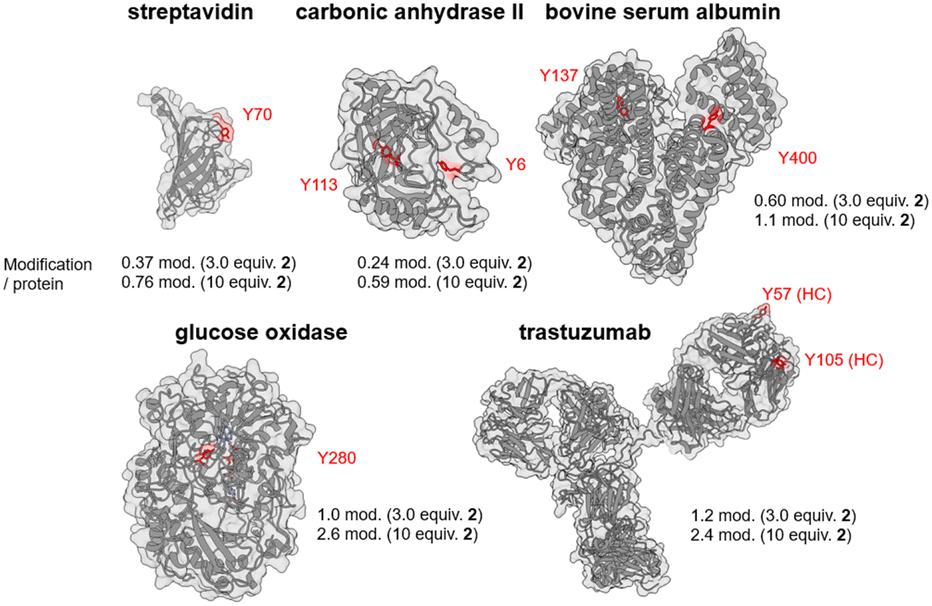 | ||
| Fig. 3 Amount of labeling per protein and major labeling sites in the Tyr click reaction using proteins as substrates. | ||
To understand the Tyr labeling by laccase, the proteome in the A431 cell lysate was labeled using MAUra-DTB (compound 8), and the labeled Tyrs were analyzed. Desthiobiotin (DTB) was selected for its elution efficiency from NeutrAvidin. Labeled proteins were digested and enriched with NeutrAvidin beads; binding sites were identified by nanoLC-MS/MS (Fig. 4a). The number of labeled proteins increased with reaction time (Fig. S51a, ESI†). Over 3000 single-modification and about 500 double-modification sites were identified (Fig. S51b and c, ESI†). In a 10 min reaction, 2017 single and 17 double-modification sites were found, indicating a preference for single modifications. In contrast, only 12 single-modification sites were identified for Trp, and no double-modification sites were detected on Trp across all reaction times. Highly reactive Tyrs (labeled at 10 min) and mildly reactive Tyrs (labeled at 60 min but not at 10 min) showed different relative accessible surface area (RSA) values, calculated using the DSSP program23 based on the predicted protein structures from the AlphaFold Protein Structure Database24 (Fig. 4c right, Fig. S52, ESI†). On the other hand, comparison of the results of motif analysis between the two groups showed that, in common, they preferred to label environments with hydrophilic amino acid residues in the vicinity (Fig. 4b). These results suggest that extremely exposed tyrosine residues are preferentially labeled at short reaction times, while moderately exposed tyrosines can also be labeled at longer reaction times. The average values of the top 100 intensities at each reaction time shown in Fig. 4c also suggest this hypothesis. The predicted RSA for total tyrosine is 0.15,25 preferentially labeling extremely exposed Tyr (e.g., average RSA of top 100 intensities at 10 min reaction time: 0.5), especially at the earlier reaction times. In addition, the self-labeling of laccase was confirmed at three Tyrs in this reaction. These labeling sites were moderately exposed and were observed only at long reaction times. They were located far from the substrate-binding site, suggesting that labeling would have little effect on enzyme activity (Fig. S53–S56, ESI†).
 | ||
| Fig. 4 Proteome-scale analysis of protein surface selectivity by identifying labeling sites. (a) Scheme for identification of labeling sites in the cell lysate. Reaction conditions: A431 cell lysate (1 mg mL−1 protein), MAUra-DTB (8) (500 μM), and laccase (82 U mL−1), in 50 mM Tris buffer pH 6.0, 37 °C, and 800 rpm. (b) Venn diagram of peptide fragments containing single-modified Tyrs at reaction times of 10 and 60 min, and motif analysis of highly reactive Tyrs. (c) Average RSA values of labeling sites with the top 100 intensities detected in the nanoLC-MS/MS analysis for each reaction time, and the box plot of highly reactive Tyrs and mildly reactive Tyrs (Fig. S52, ESI†) (*p < 0.05). | ||
In conclusion, we developed a remarkably more efficient Tyr click reaction than conventional methods using laccase. We demonstrated that this highly efficient Tyr labeling under mild reaction conditions did not require the addition of an oxidant. In enzyme-based Tyr labeling methods, tyrosinase activates tyrosine residues or phenolic structures in labeling reagents, whereas in the current method, laccase activates the labeling reagent MAUra and efficiently catalyzes Tyr labeling. Tyr labeling by tyrosinase is limited to tyrosine residues on extremely exposed N- or C-terminal tags, whereas the current method can label the exposed Tyrs of natural-type proteins. Moreover, this reaction is a useful method for the functionalization of peptides and proteins because of unlikely side reactions and negligible effects on other amino acid residues. Furthermore, this protein labeling technique can selectively label highly surface-exposed Tyrs.
K. N.: methodology and data analysis; C. F.,S. M., Z. L., T. N., and H. T.: methodology; H. N.: software; T. K., S. T., and M. I.: supervision; S. S.: supervision and writing – original draft. All authors participated in the discussion of the results and commented on the manuscript.
This study was partially supported by research grants from the JST FOREST Program (grant number: JPMJFR2005 to S. S.), Grant-in-Aid for Scientific Research (B) (23H02099 to S. S.), Naito Foundation (to S. S.), Grant-in-Aid for Transformative Research Areas (A), Biophysical Chemistry for Material Symbiosis (21H05503 to S. S.), and Scientific Research (A) (22H00436 M. I.). We thank the Cell Biology Center Research Core Facility at Tokyo Tech for the Q-Exactive mass spectrometry measurements, generous support from the FRIS CoRE, which is a shared research environment. This study was partially supported by the Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP23ama121019. We thank Dr Izumi Sakamoto, Dr Ryohei Uematsu, and Naoko Hashimoto (GlyTech, Inc.) for their help in LC-MS experiment for the labeling site analysis.
Data availability
The data (experimental procedures, characterization data, and supporting data) for this study are openly available in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- S. D. Varfolomeev, I. V. Uporov and E. V. Fedorov, Biochemistry, 2002, 67, 1099–1108 CAS
.
- S. Sato, M. Matsumura, T. Kadonosono, S. Abe, T. Ueno, H. Ueda and H. Nakamura, Bioconjugate Chem., 2020, 31, 1417–1424 CrossRef CAS PubMed
- B. X. Li, D. K. Kim, S. Bloom, R. Y.-C. Huang, J. X. Qiao, W. R. Ewing, D. G. Oblinsky, G. D. Scholes and D. W. C. MacMillan, Nat. Chem., 2021, 13, 902–908 CrossRef CAS PubMed
- C. M. Baker and G. H. Grant, Biopolymers, 2007, 85, 456–470 CrossRef CAS PubMed
- K. A. Ryu, C. M. Kaszuba, N. B. Bissonnette, R. C. Oslund and O. O. Fadeyi, Nat. Rev. Chem., 2021, 5, 322–337 CrossRef CAS PubMed
- S. Sato, K. Nakamura and H. Nakamura, ACS Chem. Biol., 2015, 10, 2633–2640 CrossRef CAS PubMed
- S. Sato, K. Nakamura and H. Nakamura, ChemBioChem, 2017, 18, 475–478 CrossRef CAS PubMed
- S. Sato, K. Nakane and H. Nakamura, Org. Biomol. Chem., 2020, 18, 3664–3668 RSC
- K. Maruyama, T. Ishiyama, Y. Seki, K. Sakai, T. Togo, K. Oisaki and M. Kanai, J. Am. Chem. Soc., 2021, 143, 19844–19855 CrossRef CAS PubMed
- S. Sato and H. Nakamura, Angew. Chem., Int. Ed., 2013, 52, 8681–8684 CrossRef CAS PubMed
- S. Sato, K. Morita and H. Nakamura, Bioconjugate Chem., 2015, 26, 250–256 CrossRef CAS PubMed
- A. M. Marmelstein, M. J. Lobba, C. S. Mogilevsky, J. C. Maza, D. D. Brauer and M. B. Francis, J. Am. Chem. Soc., 2020, 142, 5078–5086 CrossRef CAS PubMed
- J. J. Bruins, A. H. Westphal, B. Albada, K. Wagner, L. Bartels, H. Spits, W. J. H. van Berkel and F. L. van Delft, Bioconjugate Chem., 2017, 28, 1189–1193 CrossRef CAS PubMed
- M. J. Lobba, C. Fellmann, A. M. Marmelstein, J. C. Maza, E. N. Kissman, S. A. Robinson, B. T. Staahl, C. Urnes, R. J. Lew, C. S. Mogilevsky, J. A. Doudna and M. B. Francis, ACS Cent. Sci., 2020, 6, 1564–1571 CrossRef CAS PubMed
- H. Zhu, J. H. Oh, Y. Matsuda, T. Mino, M. Ishikawa, H. Nakamura, M. Tsujikawa, H. Nonaka and I. Hamachi, J. Am. Chem. Soc., 2024, 146, 7515–7523 CrossRef CAS PubMed
- M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS
- P. J. Kersten, B. Kalyanaraman, K. E. Hammel, B. Reinhammar and T. K. Kirk, Biochem. J., 1990, 268, 475–480 CrossRef CAS PubMed
- S. Depienne, D. Alvarez-Dorta, M. Croyal, R. C. T. Temgoua, C. Charlier, D. Deniaud, M. Mével, M. Boujtita and S. G. Gouin, Chem. Sci., 2021, 12, 15374–15381 RSC
- Note: The reaction rate was estimated from the rate of decrease of angiotensin II. The concentration of laccase (Amano Enzyme Inc. LC-Y120) corresponding to 2.5 U mL−1 was 13 nM (Fig. S4, ESI†).
- S. Kurniawati and J. A. Nicell, Bioresour. Technol., 2008, 99, 7825–7834 CrossRef CAS PubMed
- M. J. Bausch, B. David, P. Dobrowolski, C. Guadalupe-Fasano, R. Gostowski, D. Selmarten, V. Prasad, A. Vaughn and L. H. Wang, J. Org. Chem., 1991, 56, 5643–5651 CrossRef CAS
- H. Ban, J. Gavrilyuk and C. F. Barbas, J. Am. Chem. Soc., 2010, 132, 1523–1525 CrossRef CAS PubMed
- R. P. Joosten, T. A. H. te Beek, E. Krieger, M. L. Hekkelman, R. W. W. Hooft, R. Schneider, C. Sander and G. Vriend, Nucleic Acids Res., 2011, 39, D411–D419 CrossRef CAS PubMed
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Žídek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, Nucleic Acids Res., 2022, 50, D439–D444 CrossRef CAS PubMed
- B. Zhang, L. Li and Q. Lü, Biomolecules, 2018, 8, 33 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03802a |
| This journal is © The Royal Society of Chemistry 2024 |