
Fe–Nx sites coupled with Fe3C on porous carbon from plastic wastes for oxygen reduction reaction†
Xiaole
Jiang‡
a,
Rui
Zhang‡
a,
Qingqing
Liao
a,
Hanjun
Zhang
a,
Yaoyue
Yang
 a and
Fan
Zhang
*b
a and
Fan
Zhang
*b
aLaboratory of Fundamental Chemistry of the State Ethnic Commission, School of Chemistry and Environment, Southwest Minzu University, Chengdu 610041, China
bKey Laboratory of Green Chemistry and Technology, Ministry of Education, National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), College of Chemistry, Sichuan University, Chengdu 610065, China. E-mail: fanzhang@scu.edu.cn
First published on 26th August 2024
Abstract
Isolated Fe–Nx sites coupled with Fe3C nanoparticles co-embedded in N-doped porous carbon were fabricated using polyethylene terephthalate wastes as carbon sources. Benefiting from the synergistic effect between Fe–Nx sites and Fe3C, and the hierarchical porous structure, the catalyst exhibits outstanding ORR performance, realizing the concept of turning trash into treasure.
Nowadays, polyethylene terephthalate (PET) is extensively used in applications such as food and beverage packaging. Global PET demand is forecasted to rise to 22.65 million tonnes by 2025.1 Unfortunately, the production of PET consumes a lot of fossil resource, and the low biodegradability of PET contributes to the accumulation of plastic waste, causing severe threats to the energy demand and natural ecosystem.2–4 At the same time, the incineration of PET wastes can lead to significant air pollution.1,3 Therefore, developing sustainable and efficient strategies to recycle PET waste into value-added products is imperative. Considering the significant carbon content in plastic wastes, PET wastes have been regarded as sustainable carbon resources for synthesizing carbon nanomaterials.4–7 Tang et al. efficiently converted waste PET bottles into porous carbon nanosheets (PCNS) by combining catalytic carbonization and the KOH activation process. The obtained PCNS displayed outstanding capability in double-layer supercapacitors of 169 F g−1 in 6 M KOH.4 Zhang et al. transformed PET wastes into N-doped carbon sheets (LPNCS) as carriers to disperse Ru nanoparticles. The Ru/LPNCS displayed outstanding HER performance with a low overpotential (15 mV@10 mA cm−2).6 Despite these advances, carbon materials derived from PET wastes for oxygen reduction reaction (ORR) have rarely been reported.
Developing sustainable energy conversion systems has been regarded as one promising strategy to alleviate the energy crisis and environmental pollution.8 Zn–air batteries (ZABs) are drawing great attention owing to their high energy density, environment-friendliness, low cost, and high safety.9 However, the energy conversion efficiency of ZABs is limited by the sluggish kinetics of the cathodic ORR.10 Although platinum-group nanomaterials have demonstrated excellent ORR performance, their scarcity, high cost, and dissatisfactory stability heavily hurdled their large-scale commercial application.11
Recently, atomically dispersed metal–nitrogen–carbon (M–N–C, M = Co, Fe, Ni, etc.) catalysts have held a promising performance for ORR.12,13 However, achieving optimal catalytic activity with only isolated active sites is challenging due to the linear scaling relations between the adsorption/desorption toward complex multiple intermediate reactions.14 Noteworthy, introducing some synergistic components, such as metal nanoparticles and metal sulfides adjacent to the isolated M–Nx sites, has been proposed to regulate the adsorption/desorption energy barriers of reaction intermediates, thereby accelerating the ORR process.15–17 Chen et al. successfully constructed a coupling catalyst with Fe–N4 and FeS NPs embedded in N/S-doped porous carbon (FeS/FeNSC). The oxidized sulfur and FeS NPs synergistically modulated the electronic structure of Fe–N4 sites, leading to outstanding ORR performance and superior stability.17
Herein, isolated Fe–Nx sites coupled with Fe3C nanoparticles embedded in N-doped porous carbon were successfully fabricated by using PET wastes as a carbon source. The Fe3C/Fe–N–C catalyst possessed outstanding ORR performance, attributed to the synergistic effect between Fe3C nanoparticles and Fe–Nx sites as well as the hierarchical porous structure. Specifically, it has an onset potential (Eonset) of 0.99 V and a half-wave potential (E1/2) of 0.89 V (vs. RHE), respectively. Moreover, the Fe3C/Fe–N–C-based ZAB revealed a maximum power density of 211 mW cm−2 and long discharge stability, indicating its potential for practical energy devices. This research not only demonstrates the reuse of plastic waste but also provides an effective catalyst for fuel cell/ZAB applications, contributing to the concept of converting waste into valuable resources.
The facile synthesis process of the Fe3C/Fe–N–C catalyst is depicted in Fig. 1a. Typically, N-doped porous carbon was synthesized from the cost-effective carbon source of PET wastes via the MgO-templated method. Subsequently, the Fe-phenanthroline (Fe-Phen) was anchored on the porous carbon via the incorporation method. Finally, the Fe-Phen/N–C mixture was converted into isolated Fe–Nx sites coupled with Fe3C nanoparticles embedded in N-doped porous carbon after pyrolysis. The carbonization process of the PET mixture was investigated by thermogravimetric analysis (TGA). As shown in Fig. 1b, the pyrolysis of the PET/MgO/cyanoguanidine (DCD) mixture displays four stages. The first stage involves heating from room temperature to 208 °C with a weight loss of 4.7 wt% due to the evaporation of the DCD. The weight loss in the second and third stages (212 to 342 °C) is attributed to the decomposition of DCD.18,19 Following this, there is a 12 wt% weight loss, which corresponds to the PET carbonation and the formation of N-doped carbon.6 The XRD patterns of all the synthesized catalysts are displayed in Fig. 1c. Only two diffraction peaks at 23.9° and 43.4° belonged to the C (002) and (101) planes can be observed in N–C and Fe–N–C catalysts,20 indicative of the absence of Fe-based particles in Fe–N–C catalyst. In contrast, the XRD pattern of Fe3C/Fe–N–C exhibits additional diffraction peaks assigned to Fe3C (PDF#35-0772), implying the formation of Fe3C with increasing pyrolysis temperature.14
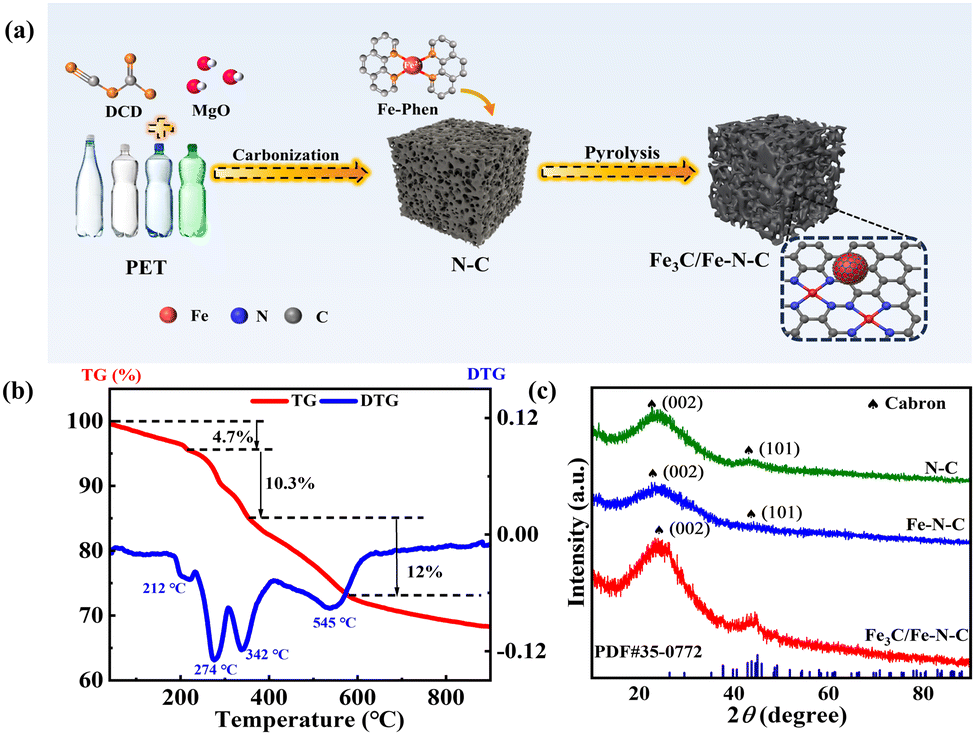 | ||
| Fig. 1 (a) Schematic illustration for the synthesis of Fe3C/Fe–N–C catalyst. (b) TG and DTG curves of PET/MgO/DCD mixture under Ar atmosphere. (c) XRD patterns of N–C, Fe–N–C, and Fe3C/Fe–N–C catalysts. | ||
The morphology and microstructure of the catalysts were observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As displayed in Fig. 2a and Fig. S1a, b (ESI†), similar 3D interconnected architecture is observed for all the catalysts, and the HRTEM images (Fig. 2b and Fig. S1c–f, ESI†) reveal that 3D carbon structure is assembled by interconnected porous nanosheets. Furthermore, the HRTEM images of the Fe–N–C catalyst (Fig. S1d and f, ESI†) confirm the absence of metal nanoparticles, which agrees with the XRD results. The HADDF-STEM images (Fig. S2, ESI†) show that abundant bright spots are uniformly dispersed in the carbon substrate, indicating that Fe species are present as isolated Fe atoms. For comparison, large amounts of iron-based nanoparticles with a size of 5–20 nm encapsulated by a few-layer graphitized carbon shell can be observed in Fe3C/Fe–N–C catalyst (Fig. 2b). Meanwhile, as displayed in Fig. S3a (ESI†), the lattice distance of the nanoparticle is 0.197 nm, corresponding to the (112) plane of Fe3C.21,22 Additionally, the HADDF-STEM images (Fig. 2c and d) reveal that abundant isolated Fe atoms (bright spots) distributed around the Fe3C nanoparticle, confirming the coexistence of atomically dispersed Fe–Nx sites and Fe3C. Moreover, energy-dispersive X-ray (EDX) elemental mappings confirm the homogeneous distribution of C, N, and Fe in the Fe3C/Fe–N–C catalyst (Fig. S3b, ESI†).
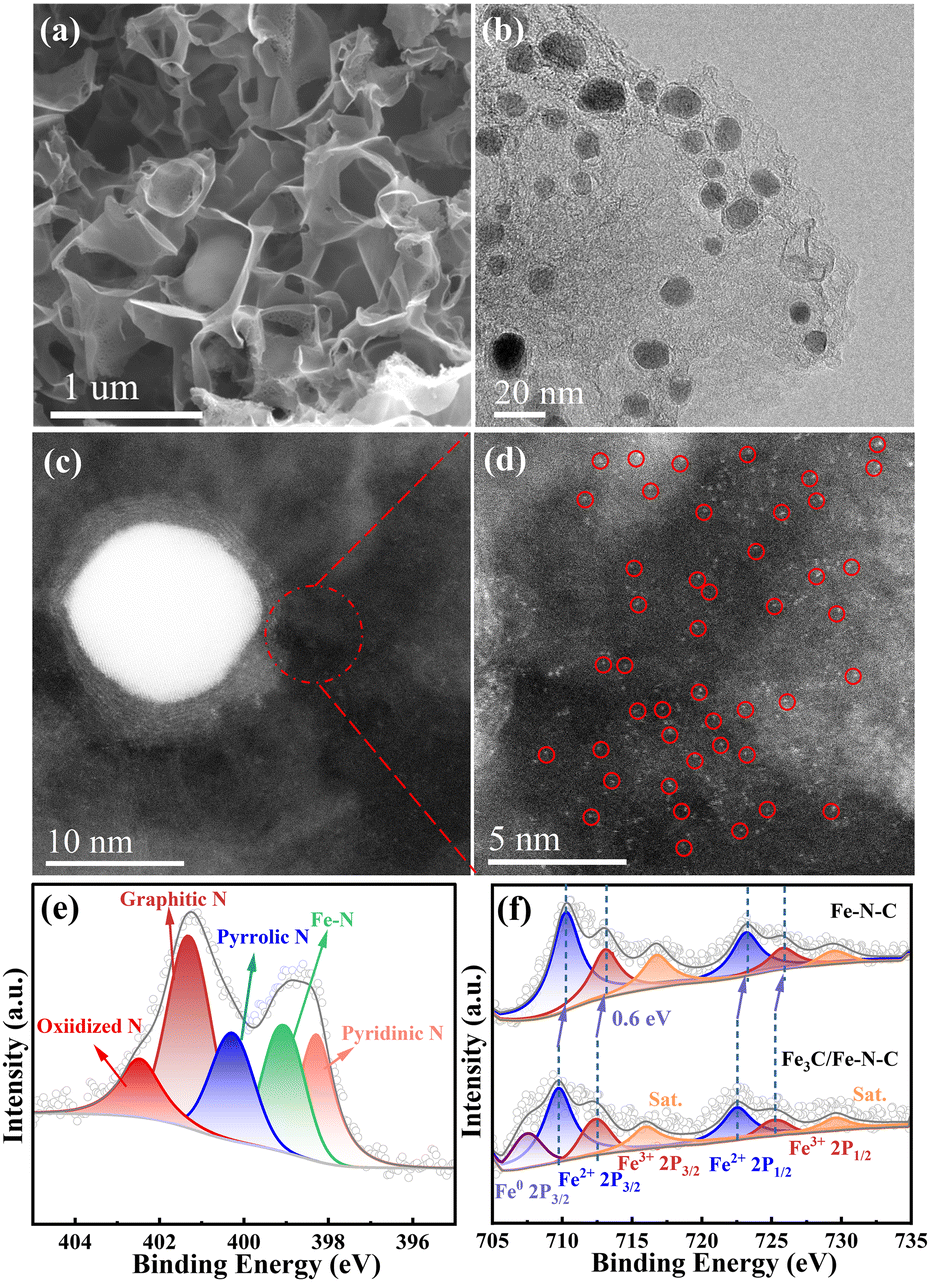 | ||
| Fig. 2 (a) SEM and (b) HRTEM images of Fe3C/Fe–N–C. (c) and (d) HAADF-STEM images of Fe3C/Fe–N–C (isolated Fe sites are highlighted by the red circles). (e) High-resolution N 1s spectra of Fe3C/Fe–N–C. (f) Fe 2p XPS spectra of the Fe–N–C and Fe3C/Fe–N–C. | ||
The Raman spectra of all the catalysts display two typical peaks at 1344 and 1581 cm−1, attributed to the defects carbon (D band) and the graphitic carbon (G band), respectively. As shown in Fig. S4 (ESI†), the D to G band intensity ratio (ID/IG) of Fe–N–C (1.08), Fe3C/Fe–N–C (1.00), and N–C (1.17) have no obvious difference.16,23 The isotherms curves of N–C, Fe–N–C, and Fe3C/Fe–N–C exhibit apparent adsorption at the low-pressure region and a typical hysteresis loop, indicating the coexistence of micropores and mesopores (Fig. S5, ESI†). The BET surface areas were calculated to be 444, 427, and 480 m2 g−1 for N–C, Fe–N–C, and Fe3C/Fe–N–C catalysts, respectively (Table S1, ESI†). Besides, the pore-size distribution reveals that mesopores with diameters ranging from 3–35 nm are predominantly presented in Fe–N–C and Fe3C/Fe–N–C. The high surface area and hierarchical porous structure would be beneficial for exposing active sites and enhancing mass transport during the ORR process.9
The surface chemical state and compositions of all the catalysts (Table S2, ESI†) were analyzed by X-ray photoelectron spectroscopy (XPS). The high-resolution N 1s spectra of all the catalysts (Fig. 2e and Fig. S6, ESI†) confirmed the presence of pyridinic-N (∼398.2 eV), Fe–N (∼399.02 eV), pyrrolic-N (∼400.03 eV), graphitic-N (∼401.05 eV) and oxidized-N (∼402.22 eV).24 The Fe 2p XPS spectrum (Fig. 2f) for Fe–N–C displays six peaks at 710.31, 723.16 eV, 712.99, and 725.79 eV, which correspond to Fe 2p1/2 and Fe 2p3/2 orbitals of Fe2+ and Fe3+ as well as a pair of satellite peaks. Notably, apart from the oxidized Fe species, a peak at 707.4 eV belonging to Fe0 (originating from Fe3C) is detected for the Fe3C/Fe–N–C catalyst.25,26 Meanwhile, the binding energies of Fe2+ and Fe3+ in Fe3C/Fe–N–C catalyst negatively shift by 0.6 eV compared with those of Fe3C/Fe–N–C, implying the electronic interactions between Fe–Nx and Fe3C nanoparticles.17,27
The ORR activities of the as-synthesized catalysts and commercial Pt/C (40 wt%) were evaluated in the O2-saturated 0.1 M KOH solution. As shown in Fig. 3a, Fe3C/Fe–N–C displays the best ORR catalytic performance. In detail, the Eonset and E1/2 are significantly enhanced from N–C to Fe–N–C catalysts, indicating that the Fe–Nx active sites boost the ORR catalytic activity. The Fe3C/Fe–N–C catalyst presents the most positive Eonset of 0.99 V (vs. RHE) and E1/2 of 0.89 V, superior to those of Fe–N–C (Eonset = 0.94 V, E1/2 = 0.86 V), and Pt/C (Eonset = 0.94 V, E1/2 = 0.83 V) catalysts. Note that the outstanding ORR activity of Fe3C/Fe–N–C compared to Fe–N–C could ascribed from the introduction of Fe3C NPs, indicating that the electronic structure of Fe–Nx sites is tuned by the Fe3C NPs and their synergistic effect significantly boosts the ORR process.15,17 Furthermore, the Fe3C/Fe–N–C catalyst presents the lowest Tafel slope (57.76 mV dec−1), and even lower than 69.42 mV dec−1 of Pt/C, implying its faster kinetics (Fig. 3b). The kinetic current density (JK) at 0.85 V of Fe3C/Fe–N–C catalyst is 23.68 mA cm−2, which is much superior to those of Fe–N–C (7.18 mA cm−2) and Pt/C (3 mA cm−2) catalysts (Fig. 3c). To investigate the reaction pathways, RRDE tests were conducted on all the catalysts. As shown in Fig. 3d, the H2O2 yield of Fe3C/Fe–N–C is below 12%, obviously smaller than those of N–C and Fe–N–C. The electron transfer number (n) of the Fe3C/Fe–N–C catalyst reaches 3.9, which is further confirmed by the number obtained from the K–L plot (Fig. S7, ESI†), indicating that the 4 e− pathway occurred on the Fe3C/Fe–N–C catalyst.28 Thus, the Fe3C/Fe–N–C catalyst outperforms all other reported catalysts derived from plastic wastes and is comparable to other state-of-the-art Fe-based ORR catalysts (Fig. 3e and Table S3, ESI†).
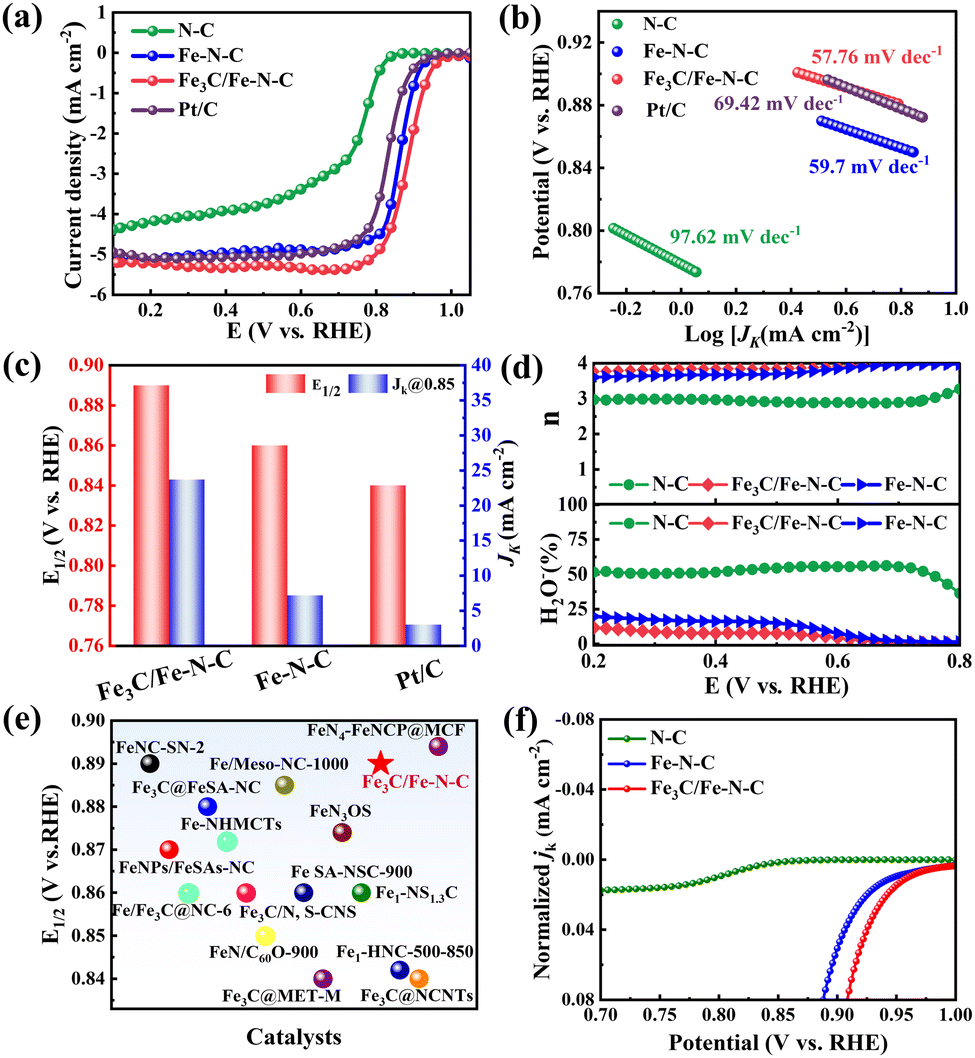 | ||
| Fig. 3 (a) LSV curves of different catalysts in O2 saturated 0.1 M KOH. (b) Tafel slopes of different catalysts. (c) E1/2 and JK of all the catalysts. (d) The H2O2 yields and electron transfer numbers of the catalysts. (e) Based on E1/2, performance comparison of different catalysts. (f) Normalized JK by ECSA for N–C, Fe–N–C, and Fe3C/Fe–N–C. | ||
The durability and methanol tolerance are significant for the practical application of electrocatalysts. As depicted in Fig. S8 (ESI†), the Fe3C/Fe–N–C catalyst exhibits 82% retention of the initial current after 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, while the Pt/C catalyst retained only 70%. Furthermore, the Fe3C/Fe–N–C catalyst maintains a stable current after the injection of methanol into the electrolyte. In contrast, the current of the Pt/C significantly decreases, indicative of the excellent methanol tolerance of the Fe3C/Fe–N–C catalyst. These results suggest that Fe3C/Fe–N–C catalyst exhibits promising potential for applications in fuel cells.
000 s, while the Pt/C catalyst retained only 70%. Furthermore, the Fe3C/Fe–N–C catalyst maintains a stable current after the injection of methanol into the electrolyte. In contrast, the current of the Pt/C significantly decreases, indicative of the excellent methanol tolerance of the Fe3C/Fe–N–C catalyst. These results suggest that Fe3C/Fe–N–C catalyst exhibits promising potential for applications in fuel cells.
To gain deep insights into the origin of excellent ORR performance, the electrochemical active surface area (ECSA) is calculated from the double-layer capacitance (Cdl), which is obtained from the cyclic voltammetry curves in the non-faradaic potential region.24 As shown in Fig. S9 (ESI†), the Cdl values of N–C, Fe–N–C and Fe3C/Fe–N–C are 20.5, 9.3 and 12.2 mF cm−2, respectively. Furthermore, the JK of all the catalysts was normalized by the ECSA. The normalized JK curves consistently showed a similar trend to the LSV curves. This suggests that the enhanced catalytic activity of Fe–N–C and Fe3C/Fe–N–C compared to N–C is primarily attributed to the improved intrinsic activity stemming from the Fe species rather than the effect of the electrochemical active surface area.17 As it is widely recognized that SCN− can deactivate the Fe–Nx sites, we conducted a KSCN poisoning experiment to confirm the active sites.15 As depicted in Fig. S10 (ESI†), the E1/2 of Fe–N–C and Fe3C/Fe–N–C significantly decrease after adding 20 mM KSCN into the electrolyte, suggesting that the isolated Fe–Nx sites are the main ORR active sites, which agrees with the previous reports.13,15 Combined with the above results, the coexistence of Fe3C NPs and atomically dispersed Fe–Nx sites play an important role in improving the ORR performance of Fe3C/Fe–N–C catalyst.
To further explore the practical applications of the Fe3C/Fe–N–C catalyst, we assembled a ZAB using catalyst-loaded carbon paper as the cathode, zinc foil as the anode, and a 6 M KOH with 0.2 M zinc acetate solution as the electrolyte, respectively. As exhibited in Fig. 4, the open-circuit potential of Fe3C/Fe–N–C based ZAB is 1.5 V, which is higher than that of the Pt/C-based counterpart (1.44 V). Additionally, the Fe3C/Fe–N–C-based ZAB delivers a maximum power density of 211 mW cm−2, outperforming the Pt/C-based ZAB (174 mW cm−2) and most of the reported carbon-based catalysts (Table S3, ESI†). The Fe3C/Fe–N–C based battery displays a stable discharge platform during 100 hours at 10 mA cm−2, indicating the outstanding stability of Fe3C/Fe–N–C catalyst for practical application in ZAB (Fig. 4b). Meanwhile, our ZAB could provide sufficient power to energize a light-emitting diode (Fig. 4b).
 | ||
| Fig. 4 (a) Discharging polarization and power density curves of Fe3C/Fe–N–C and Pt/C. (b) Corresponding discharging curves at 10 mA cm−2 of the Fe3C/Fe–N–C based ZAB (photograph of an LED panel lighted up by Fe3C/Fe–N–C based ZAB). | ||
In summary, a simple and facial strategy has been proposed to synthesize atomically dispersed Fe–Nx sites and Fe3C nanoparticles coexistence in N-doped porous carbon (Fe3C/Fe–N–C) by using low-cost PET waste as the carbon precursor. The Fe3C/Fe–N–C catalyst exhibited outstanding ORR performance and superior durability in alkaline media, owing to the synergistic effect between Fe–Nx sites and Fe3C nanoparticles, as well as the hierarchical porous structure. Our work not only offers an effective way to recycle PET wastes into “gold electrocatalyst” but also sheds light on the precise design of high-performance ORR catalysts with tunable electronic configurations.
This work was financially supported by the National Key R&D Program of China (2021YFA1501700), the National Science Foundation of China (22272114), Open Project Fund of State Key Laboratory of Catalysis (No. N-22-09), Fundamental Research Funds from Sichuan University (2022SCUNL103) and the Funding for Hundred Talent Program of Sichuan University (20822041E4079).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Manjunathan, K. Pavithra, S. Nangan, S. Prakash, K. K. Saxena, K. Sharma, K. Muzammil, D. Verma, J. R. Gnanapragasam, R. Ramasubburayan and M. Revathi, Chemosphere, 2024, 353, 141541 CrossRef CAS PubMed.
- R. Nistico, Polym. Test., 2020, 90, 106707 CrossRef CAS.
- S. Shariffan and N. Asasian-Kolur, J. Anal. Appl. Pyrolysis, 2022, 163, 105496 CrossRef.
- Y. L. Wen, K. Kierzek, J. K. Min, X. C. Chen, J. Gong, R. Niu, X. Wen, J. Azadmanjiri, E. Mijowska and T. Tang, J. Appl. Polym. Sci., 2020, 137, 48338 CrossRef CAS.
- J. Gong, X. Chen and T. Tang, Prog. Polym. Sci., 2019, 94, 1–32 CrossRef CAS.
- C. Juan, B. Lan, C. Zhao, H. Zhang, D. Li and F. Zhang, Chem. Commun., 2023, 59, 6187–6190 RSC.
- F. Zhang, F. Wang, X. Y. Wei, Y. Yang, S. M. Xu, D. H. Deng and Y. Z. Wang, J. Energy Chem., 2022, 69, 369–388 CrossRef CAS.
- H. J. Huang, D. S. Yu, F. Hu, S. C. Huang, J. N. Song, H. Y. Chen, L. L. Li and S. J. Peng, Angew. Chem., Int. Ed., 2022, 61, e202116068 CrossRef CAS PubMed.
- X. N. Tang, Y. H. Wei, W. J. Zhai, Y. G. Wu, T. Hu, K. Yuan and Y. W. Chen., Adv. Mater., 2023, 35, 2208942 CrossRef CAS PubMed.
- J. Q. Bermejo, A. Daouli, S. G. Dalí, Y. D. Cui, A. Zitolo, J. C. Gutiérrez, M. Emo, M. T. Izquierdo, W. Mustain, M. Badawi, A. Celzard and V. Fierro, Adv. Funct. Mater., 2024, 2403810 CrossRef.
- Y. L. Zhao, Z. B. Tian, W. Q. Wang, X. H. Deng, J. C. Tseng and G. H. Wang, Green Carbon, 2024, 2, 221–230 CrossRef.
- C. Z. Zhu, Q. R. Shi, B. Z. Xu, S. F. Fu, G. Wan, C. Yang, S. Y. Yao, J. H. Song, H. Zhou, D. Du, S. P. Beckman, D. Su and Y. H. Lin, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- X. l Jiang, Y. Y. Yang, C. l Zhu, C. Zhou, R. Zhang, F. Wu, H. H. Wu and J. Wang, Int. J. Hydrogen Energy, 2022, 47, 33308–33318 CrossRef CAS.
- J. W. Chang, Q. Zhang, J. K. Yu, W. Jing, S. Y. Wang, G. C. Yin, G. I. N. Waterhouse and S. Y. Lu, Adv. Sci., 2023, 10, e2301656 CrossRef PubMed.
- W. J. Zhai, S. Huang, C. Lu, X. Tang, L. Li, B. Huang, T. Hu, K. Yuan, X. Zhuang and Y. Chen, Small, 2022, 18, 2107225 CrossRef CAS PubMed.
- H. Liu, L. Z. Jiang, Y. Y. Sun, J. Khan, B. Feng, J. M. Xiao, H. D. Zhang, H. J. Xie, L. N. Li, S. Y. Wang and L. Han, Adv. Energy Mater., 2023, 13, 2301223 CrossRef CAS.
- J. D. Chen, B. Y. Huang, R. Cao, L. B. Li, X. N. Tang, B. Wu, Y. G. Wu, T. Hu, K. Yuan and Y. W. Chen, Adv. Funct. Mater., 2022, 33, 2209315 CrossRef.
- Z. Z. Lu, D. Liu, J. T. Zhou and F. Dong, Chem. Eng. J., 2017, 314, 640–649 CrossRef CAS.
- J. B. Zhang, Z. C. Tan, S. H. Meng, S. H. Li and L. M. Zhang, Thermochim. Acta, 1997, 3, 11–15 Search PubMed.
- X. Y. Lu, Y. Q. Li, D. Dong, Y. B. Wan, R. P. Li, L. H. Xiao, D. Wang, L. L. Liu, G. Z. Wang, J. Q. Zhang, M. An and P. Yang, J. Colloid Interface Sci., 2024, 653, 654–663 CrossRef CAS PubMed.
- H. G. Zhang, Y. Wang, T. Wu, J. X. Yu, S. R. B. Arulmani, W. T. Chen, L. Su, M. Huang, J. Yan and X. J. Liu, J. Alloy. Compd., 2023, 944, 169039 CrossRef CAS.
- R. Q. Zhong, Y. X. Wu, Z. B. Liang, W. H. Guo, C. X. Zhi, C. Qu, S. Gao, B. J. Zhu, H. Zhang and R. Q. Zou, Carbon, 2019, 142, 115–122 CrossRef CAS.
- C. Liu, J. Zheng, B. Chi, C. Zhong, Y. Deng, C. Chen, D. Dang, W. Fan, Z. Cui and Q. Liu, Energy Environ. Sci., 2024, 17, 5941–5949 RSC.
- Q. L. Ye, M. H. Li, T. Y. Xiao, S. Y. Hou, Y. J. Deng, J. M. Luo and X. L. Tian, Int. J. Hydrogen Energy, 2024, 49, 1014–1021 CrossRef CAS.
- X. Cui, L. K. Gao, S. Lei, S. Liang, J. W. Zhang, C. D. Sewell, W. D. Xue, Q. Liu, Z. Q. Lin and Y. K. Yang, Adv. Funct. Mater., 2020, 31, 2009197 CrossRef.
- Y. H. Liu, X. K. Wang, B. L. Zhao, X. Shao and M. H. Huang, Chem. – Eur. J., 2019, 25, 9650–9657 CrossRef CAS PubMed.
- C. H. Zhang, X. K. Wang, K. Song, K. Y. Chen, S. X. Dai, H. L. Wang and M. H. Huang, Nano Res., 2023, 16, 9371–9378 CrossRef CAS.
- S. X. Li, G. Y. Xing, S. Zhao, J. Peng, L. F. Zhao, F. Hu, L. L. Li, J. Z. Wang, S. Ramakrishna and S. J. Peng, Natl. Sci. Rev., 2024, 11, nwae193, DOI:10.1093/nsr/nwae193.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03683b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |