
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into coordination-cage based catalysis
Michael D.
Ward
 *
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: m.d.ward@warwick.ac.uk
First published on 29th August 2024
Abstract
This review article summarises work from the author's group on catalysis using coordination cages over the (approximate) period 2018–2024. Recent insights discussed include (i) the general mechanism of catalysis, which involves co-location of reaction partners using orthogonal interactions involving the cage cavity (neutral hydrophobic substrates) and the surface anion-based reaction partners; (ii) the role of the cage exterior surface in facilitating catalysis in some cases; (iii) quantitative analysis of anion-binding to the cage surface, as a complement to measurement of binding constants of neutral guests inside the cavity; (iv) a new type of redox-based catalysis using reactive oxygen species, which are generated by reaction of oxidants such as H2O2 and HSO5− with Co(II)/Co(III) redox couples in the cage superstructure. Collectively the results discussed provide signficant new possibilities for further exploration of catalysis using supramolecular assemblies.
1. Introduction and background
The chemistry of coordination cages remains an area of fascination for a combination of several reasons.1 These include (i) the fascination with their high-symmetry, elaborate structures which can arise from self-assembly processes based on very simple component parts; (ii) their ability to bind small molecule guests inside the central cavities; and (iii) functional behaviour which arises from guest binding, such as transport, drug delivery, and catalysed reactions of bound guests.1 It is this last area that is the subject of this review article, which provides a summary of some new directions in cage-based catalysis from this group over the last few years. Catalysis in coordination cages has been an immensely popular and productive field recently with reviews on different aspects of this published by (amongst others) the groups of Raymond and Toste;1j,1m,1p Verpoort;1e Nitschke;1f,1t Zhou;1i Fujita;1k Otte;1l Jin;1n He;1o Lusby;1q Duan;1r and Reek.1sIn 2018 we published a review article summarising work over two decades in the self-assembly, host–guest chemistry and catalysis properties of a family of polyhedral coordination cages based on ditopic and tritopic ligands bearing chelating pyrazolyl–pyridine ligating units.2 A wide range of coordination cages had been prepared and structurally characterised: and the host–guest chemistry of an octanuclear M8L12 cage family (denoted H, Hw or Hpeg according to the nature of external substituents appended for solubility reasons; see Fig. 1) with an approximately cubic shape,3 whose size and high stability lent itself well to binding small-molecule guests in a range of solvents,4 was studied in detail, leading to a well-developed understanding of the structural and thermodynamic factors underpinning guest recognition. This understanding was (and is) sufficiently well developed that it could be used as the basis of a computational tool to predict guest binding strengths in one particular host cage in water.5
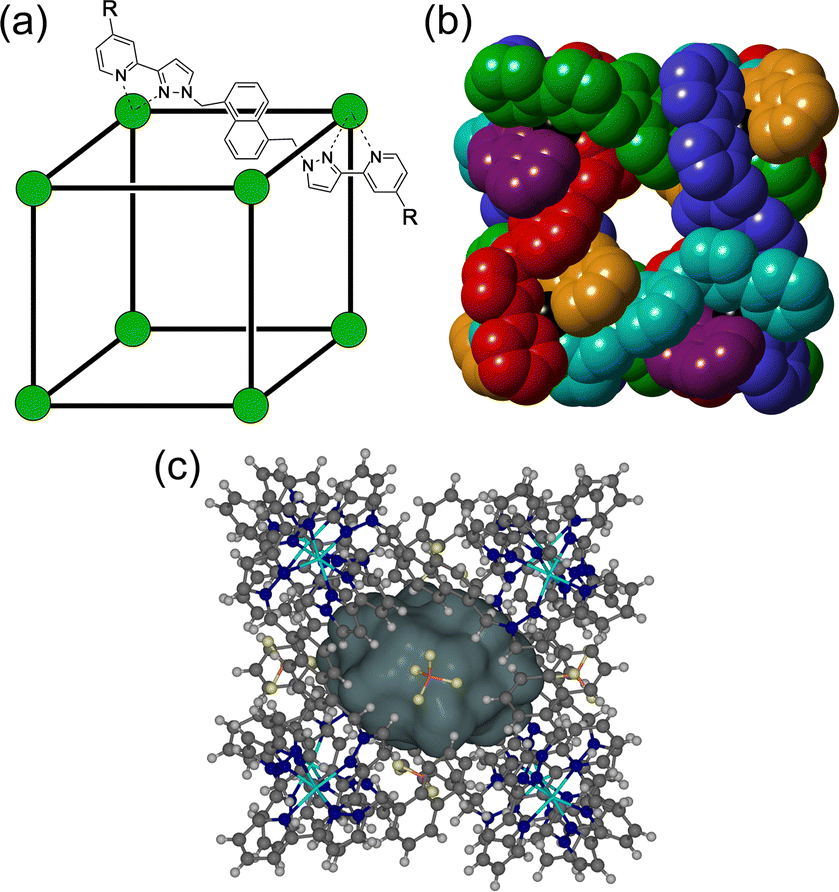 | ||
| Fig. 1 (a) Cartoon illustrating the cubic host cage [M8L12]16+, abbreviated as H (R = H), emphasising the cubic array of Co(II) ions and the disposition of one bridging ligand; and its derivatives bearing substituents at the twenty-four externally-directed pyridyl C4 positions Hw (R = CH2OH), and Hpeg [R = –(CH2OCH2)3CH2OMe]. (b) A view of the complete structure with all ligands shown, coloured differently to emphasise their overall disposition and the presence of portals in the faces. (c) A view of the complete cage structure, highlighting the guest binding cavity space (V = 409 Å3). | ||
A particularly notable result from this work was the observation of very efficient catalysis of a reaction of a cavity-bound guest, viz. the >105-fold rate enhancement (kcat/kuncat) of the Kemp elimination reaction of cavity-bound benzisoxazole in weakly basic conditions.6 A key factor contributing to this catalysis turned out to be the accumulation of anions in aqueous solution around the surface of the M8L12 cage, which carries a 16+ charge due to the use of first-row M2+ cations, usually Co(II). The windows defined by the ligand array in the square faces of the M8L12 assembly through which guests can exchange between bulk solution and cage cavity are, fortuitously, the right size to accommodate a range of anions which form a large number of CH⋯X− hydrogen bonds. This results in a high (ca. 0.1 M) local concentration of surface-bound anions surrounding the cavity-bound guest, and the catalysis can be ascribed to this co-location of reaction partners which are brought together by orthogonal interactions, with the hydrophobic effect responsible for binding benzisoxazole inside the cage cavity, and an electrostatic/ion-pairing effect responsible for the accumulation of hydroxide ions around it. The 105-fold catalytic rate enhancement of the Kemp elimination can be ascribed simply to the fact that even at a bulk pH which is modest (8–9), having hydroxide ions in all six windows surrounding the cavity-bound guest results in a local pH around the guest in the range 13–14:6 and the Kemp elimination is first order in base concentration.
This observation prompted us to study cage-based catalysis with our cage family in more detail, and this article provides an update which summarises advances in several key areas made in the last few years. These are (i) the study of additional examples of cage-catalysed hydrolysis, including the location of the catalysed reaction (inside the cavity, or outside the cavity at the external surface); (ii) further work on host/guest chemistry, and in particular the number of guests binding inside the cage cavity and the effect that this can have on catalysis; (iii) a detailed study of interactions of anions with the M8L12 cage surface, and how this contributes to catalysis; and (iv) recent observations of the role of cage-based redox activity in mediating some types of catalytic reaction.
2. Additional examples of cage-based catalysis: inside or outside the cage?
The basic catalysis mechanism – co-location of a hydrophobic substrate which interacts with the cage, and hydroxide ions which cluster around the cage due to its high positive charge2,6 – immediately prompted additional studies to look at possibilities of cage-catalysed hydrolysis reactions, with some unexpected results relating to the precise location of the catalytic reaction.The substrate ‘dichlorvos’ (2,2-dichlorovinyl dimethyl phosphate) is an appealing target for cage-catalysed hydrolysis.7 It binds (albeit weakly) in the cage cavity, and the cage/guest complex was structurally characterised (Fig. 2), showing the usual hydrogen-bonding interactions between the Lewis basic sites on the guests and the array of C–H(δ+) donors lying close to the dicationic fac tris-chelate metal centres. In addition it is chemically significant as an example of a highly toxic organophosphate insecticide (an acetylcholinesterase inhibitor): and hence of particular interest as something for which catalysed destruction could be useful.
 | ||
| Fig. 2 Crystal structure of the H•dichlorvos complex. (a) A view of the complete cage with the guest shown in space-filling mode; (b) a view of the guest (plus a MeOH molecule) emphasising the CH⋯O interactions with the cage interior surface (green dotted lines). | ||
In weakly basic conditions (pD 7.7 for NMR experiments in D2O, pH 8.5 for UV/vis experiments in H2O), a clear acceleration of the rate of dichlorvos hydrolysis was observed which was first order in Hw. Surprisingly however the control experiment – blocking the cage cavity with the very strongly-binding guest cycloundecanone, which is both highly hydrophobic and an ideal size match4b – made no difference to the reaction rate: this is in notable contrast to the original studies of the Kemp elimination, in which blocking the cage cavity with the same inert inhibitor prevented the catalysis.6 Thus it is not necessary for dichlorvos to be inside the cage to undergo a catalysed reaction with hydroxide, and the obvious conclusion is that catalysis can occur at the cage external surface: as the exterior surface of the cage is just as hydrophobic as the interior surface there can still be association with a hydrophobic substrate such as dichlorvos in solution, and the dichlorvos will thereby be brought into the vicinity of the hydroxide-rich layer, facilitating catalysis. The low binding constant (difficult to estimate accurately from a spectroscopic titration given that the catalysis happens during the titration experiment, but likely ≈ 30 M−1) means that under the experimental conditions very little dichlorvos will be cavity-bound.
We established that this was a general phenomenon by (i) demonstrating exactly similar behaviour with other organophosphate substrates such as 2- and 4-nitrophenyl-dimethylphosphate (ortho and para-oxon), noting that the latter of these does not bind in the cavity of the M8L12 cages for steric reasons, yet catalysis still occurs; and (ii) demonstrating cage-based catalysed hydrolysis of the same substrates using a much smaller M4L6 tetrahedral cage with a similar type of hydrophobic surface but no possibility of binding these guests in its tiny central cavity. In all cases second-order rate constants for the catalysis (first order in cage and first order in substrate) were in the range 10−3–10−2 M−1 s−1.7
It is interesting to reflect that most of the attention paid to coordination cages as hosts assumes that the guest will be cavity-bound: but this is clearly not essential for catalysis, although the one definitive example of cavity-based catalysis (Kemp elimination) demonstrated much more impressive rate accelerations due to the substrate being completely surrounded by the hydroxide ions bound in the cage windows. External-surface binding is therefore predictably far less effective, but – as it does not rely on the cavity – is also far more general with little in the way of shape/size restrictions that a cavity would impose. In general any surface that combines the characteristics of being both hydrophobic and cationic could bring hydrophobic substrates and hydroxide ions into proximity around the cage surface for this type of catalysis.
An additional example of this type of exterior-surface catalysis is provided by hydrolysis of diacetyl fluorescein (Fig. 3).8 This neutral, hydrophobic substrate is too large for cavity binding in the M8L12 host, but nonetheless its hydrolysis to fluorescein at pH 7 – easily followed by UV/vis spectroscopy – is substantially accelerated in the presence of Hw with a second-order rate constant for the catalysed reaction of 1 × 10−2 M−1 s−1 (comparable to what was observed for the catalysed organophosphate hydrolysis reactions).7
 | ||
| Fig. 3 (a) Hw-catalysed hydrolysis of diacetyl fluorescein; (b) dependence of the reaction rate on substrate concentration, fitted to a Michaelis Menten reaction model to extract an association constant for the interaction between substrate and cage exterior surface (see main text). | ||
The plateauing in the rate of catalysis as substrate concentration is increased is indicative of a strong cage/guest interaction which saturates the cage external surface at high substrate concentrations (Fig. 3b). This allows a Michaelis–Menten model to be used to extract a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding constant of 1.5(2) × 104 M−1 for diacetyl-fluorescein to the cage external surface, which is stronger than many cavity-binding guests and implies good host/guest contact over a large hydrophobic surface area. This strong interaction means that even under the dilute conditions appropriate for UV/vis spectroscopic analysis a significant fraction of the guest is in contact with the cage and thereby brought in to the hydroxide-rich layer around the cationic surface: being able to quantify this allowed determination of the value of kcat/kuncat (ratio of the rate constant for substrate hydrolysis in contact with the cage surface compared to free in solution under the same conditions) to be 50;9 which is significant but may be compared with a kcat/kuncat ratio of 105 for a reaction occurring inside the cavity.6 This potentially general external-surface catalysis was identified with various other substrates and reaction types, including a Kemp elimination of 5-nitro-1,2-benzisoxazole to give 2-cyano-4-nitrophenolate,9 an aldol condensation with indane-1,3-dione to give bindone,10 and some simple ester hydrolyses of carboxylate, sulfonate or sulfite esters.8,11
:1 binding constant of 1.5(2) × 104 M−1 for diacetyl-fluorescein to the cage external surface, which is stronger than many cavity-binding guests and implies good host/guest contact over a large hydrophobic surface area. This strong interaction means that even under the dilute conditions appropriate for UV/vis spectroscopic analysis a significant fraction of the guest is in contact with the cage and thereby brought in to the hydroxide-rich layer around the cationic surface: being able to quantify this allowed determination of the value of kcat/kuncat (ratio of the rate constant for substrate hydrolysis in contact with the cage surface compared to free in solution under the same conditions) to be 50;9 which is significant but may be compared with a kcat/kuncat ratio of 105 for a reaction occurring inside the cavity.6 This potentially general external-surface catalysis was identified with various other substrates and reaction types, including a Kemp elimination of 5-nitro-1,2-benzisoxazole to give 2-cyano-4-nitrophenolate,9 an aldol condensation with indane-1,3-dione to give bindone,10 and some simple ester hydrolyses of carboxylate, sulfonate or sulfite esters.8,11
The comparison of the fast cavity-based Kemp elimination of unsubstituted 1,2-benzisoxazole, with the much slower external surface-based reaction of 5-nitro-1,2-benzisoxazole (for which the second-order rate constant for catalysis is two orders of magnitude smaller), is interesting as it reveals the importance of guest orientation.9 Unsubstituted 1,2-benzisoxazole – according to the crystal structure of the cage/guest complex – can adopt a geometry in the cavity whereby the reactive CH proton whose removal is the first step of the elimination reaction is oriented towards a window in one of the cage faces where a hydroxide ion binds, and is therefore accessible to a surface-bound anion.6 In contrast, with 5-nitro-1,2-benzisoxazole as guest, the presence of additional hydrogen-bonding interactions with the cage interior surface involving the nitro substituent (again, according to the crystal structure) re-orients the guest and means that the isoxazole C–H proton is not so accessible to the array of surface-bound hydroxide ions.9 Clearly one should not extrapolate too far from solid-state structures to solution behaviour, but this accessibility (or not) of the reactive C–H proton to the hydroxide ions around the cage surface may be a significant factor in the catalytic activity, in particular whether catalysis occurs efficiently inside the cavity or less efficiently at the external surface.
The logical limit of the behaviour described above – that catalysis in many cases occurs externally but not internally, possibly due to guest orientation within the cavity and hence accessibility to the surface-bound anions – is that reaction of a substrate with hydroxide ions does not occur at all inside the cavity, an example of negative catalysis. This means that the host cage will act as a protecting group and reduce the reaction rate even compared to the uncatalyzed background reaction in the absence of cage. We observed this with 4-nitrophenylacetate as guest (K for cavity-binding = 3.5(3) × 103 M−1).8 In the presence of Hw the rate of ester hydrolysis decreases slightly, as some of the substrate is bound in the cage cavity and thereby protected. In the presence of the strongly-binding competing guest cycloundecanone which blocks the cavity,4b all of the 4-nitrophenylacetate is displaced and sits outside the cavity, and the reaction rate is slightly accelerated compared to the background reaction due to the external surface catalysis. In such a case the reaction rate constant will be concentration dependent, as the balance of positive and negative catalysis contributions will change according to what fraction of the substrate is cavity-bound (negative catalysis) and what fraction is free (positive catalysis at the external surface).8
An interesting potential application of this is the protection of reactive samples of toxic materials for forensic analysis.12O,O′-Diisopropyl fluorophosphate (DFP) is a simulant of the G-series chemical warfare agent ‘GB’ (sarin), and its decomposition by reaction with hydroxide ions in aqueous solution is substantially slowed down by its interaction with the host cage Hpeg (a PEG-ylated analogue of Hw; see Fig. 1). Crystallographic analysis (Fig. 4) shows that DFP binds in the windows around the cage surface rather than the central cavity in the solid state, but in solution the presence of the cavity-blocking competitor cycloundecanone prevents DFP from interacting with the cage, implying that protective effect/negative catalysis arises from cavity binding of DFP inside Hpeg. An exactly similar effect was observed using sarin (not a simulant!) – whose hydrolysis rate in buffered aqueous solution was substantially slowed in the presence of Hpeg: one experiment showed 58% of a sample of sarin remaining intact and un-hydrolysed after 77 minutes in the presence of Hpeg, compared to just 7% remaining in the control experiment with no cage present.12
 | ||
| Fig. 4 Crystal structure of H•DFP with the Cl− anions (dark green) and DFP guests in the windows visible. | ||
3. Crystal sponge experiments: binding of one guest or two, and the implications for catalysis
The vast majority of guest molecules for which binding in the M8L12 cage has been characterised in solution show spectroscopic titration data consistent with formation of 1:1 species. For example, spectroscopic signatures such as fluorescence quenching as a guest binds to (and is quenched by) the cage can be fitted to 1:1 binding isotherms to provide a standard binding constant. Basically all of the early crystal structures of cage/guest complexes that we reported in the 2018 review showed the presence of one guest in the cage cavity.2 The sole exception was the guest dimethyl methylphosphonate, a simple chemical warfare agent structural simulant, which is small enough for two to bind in the cage cavity, anchored by hydrogen-bonding interactions at diametrically opposite corners of the cavity.13
Our program of crystallographic analyses of cage/guest complexes was transformed by the high-throughput possibilities offered by (i) the use of the ‘crystal sponge’ method, pioneered by Fujita's group,14 which allowed generation of cage/guest crystals in large numbers; and (ii) use of synchrotron radiation coupled to the automated sample-change capabilities at the diamond facility (beamline I-19).15 Large numbers of single crystals of the M8L12 host cage H could be prepared in a single batch by a solvothermal reaction followed by slow cooling. Then, immersion of a crystal in a concentrated MeOH solution of a potential guest (or in the guest as a pure oil) for a few hours allowed guest uptake: sometimes into the central cavity, sometimes in the spaces between cage molecules such that the guest was in contact with the cage exterior surface, and sometimes both, with guests occupying both internal and external (with respect to the cavity) sites. Crystals were pre-mounted and stored under liquid N2 before analysis, and with a complete data collection plus sample changeover time taking about half an hour, 24 hours on the I-19 beamline at the Diamond synchrotron allowed >40 crystals to be evaluated of which we found typically that around one third showed successful uptake of the guest into the cage cavity giving a well-defined crystal structure. One 24-hour remote-access session therefore provided more structures of host/guest complexes than had been possible in the previous 5 years with a standard laboratory diffractometer.
We quickly found that many bulky or irregularly-shaped guests formed 1:1 host:guest complexes with H, as expected: but many near-planar guests such as simple substituted aromatics, coumarins, and substituted naphthalenes (amongst others) form H•G2 adducts with the guests bound as a centrosymmetric stacked pair, separated by typical π-stacking distances.16 Some representative examples are in Fig. 5. Invariably Lewis-basic parts of the guests (often carbonyl functional groups) were anchored in the cavity by participation in H-bonding interactions with an array of inwardly-directed C–H donors from the cage interior surface, situated close to the M2+ vertices such that CH⋯O interactions are slightly charge-assisted.17 The presence of a pair of guests could result in unexpectedly high cavity occupancies, with the guest pair occupying a proportion of the cavity space far in excess of the optimal value of 55(±9)% identified by Rebek:18 the highest packing coefficient we observed was 87% with a pair of 4-methyl-7-aminocoumarin guests.16a Whilst such high cavity occupancies in host–guest assemblies are unusual they are clearly facilitated by (a) hydrogen-bonding interactions between the guest and the cage interior surface, and (b) π–π stacking interactions between the guest pair, both of which will tend to make the guest pair occupy less space than it would otherwise.
 | ||
| Fig. 5 Crystal structures of some cage/guest complexes using cage H, with as guest (a) tetrachloro-1,2-benzoquinone; (b) di(2-pyridyl)-disulfide; (c) a stacked pair of coumarin molecules (across an inversion centre); (d) a stacked pair of 2-quinolinone molecules (across an inversion centre); and (e) a stacked pair of 4-methyl-7-amino-coumarin molecules (across an inversion centre). Views (a)–(c) show a complete cage with the guest or guests shown in space-filling view; views (d)–(e) emphasise how the stacked pair of guests interacts with the cage interior surface through a network of CH⋯O interactions (green dotted lines). | ||
The picture given by these crystallographic studies – that the cavity could accommodate guest pairs sometimes – contradicted the solution studies whereby guest binding constants are determined via NMR or fluorescence titrations; such experiments always gave titration data that fitted to a 1:1 binding model.2 This difference is not hard to rationalise. Solution studies are performed under dilute conditions where 1:1 H•G species will be more prevalent than 1:2 H•G2 species, assuming that the second binding constant K2 is smaller than the first binding constant K1, which will normally be the case unless cooperative binding is present (see later). Thus we expect that, during a spectroscopic titration to measure a solution binding constant, any H•G2 species will be present only in small amounts compared to H•G – unless an artificially large excess of guest is present and/or concentrations are high.16a In contrast the H•G2 species formed in the solid state from crystalline sponge experiments are not thermodynamic minima, but are prepared under forcing conditions, by soaking crystals containing ‘empty’ cages (actually containing disordered solvent molecules) in a very large excess of the relevant guest.
Thus the solution measurements and the crystallography measurements are complementary to one another and illustrate different aspects of the host/guest behaviour. Knowing from crystallography experiments that a particular guest can bind as a pair under crystalline sponge conditions allowed us to search for evidence of 1:2 H•G2 complex formation in solution by using either 1H NMR experiments at high guest concentrations, or Job plot experiments under carefully selected conditions where the spectroscopic changes associated with the first and then the second guest binding (Δ1 and Δ2) are necessarily equivalent and additive – an essential precondition for such measurements to give meaningful results.19a Without the knowledge that formation of 1:2 H•G2 complexes was possible at all from the crystallography experiments,16a they would never have been identified in solution, with standard titrations only revealing the 1:1 binding which dominates under normal dilute conditions. The importance of relying on a combination of complementary measurements to give a full picture of host/guest behaviour is clear, and has been the subject of reviews elsewhere.19
This behaviour turned out to be significant in a new example of cage-based catalysis: the SNAr reaction of 2,4-dinitrofluorobezene (DNFB) with hydroxide to generate 2,4-dinitrophenolate (DNP), which revealed some unexpected subtleties in its behaviour arising from the ability of both substrate DNFB and product DNP to bind inside the cavity, not just on their own but in combination.20 A key observation is that two equivalents of DNP product can be observed to bind inside the cavity in a crystal structure of the H•(DNP)2 complex: clearly the negative charge of DNP does not prevent formation of this stacked pair, no doubt because of the extensive array of charge-assisted hydrogen-bonds between the interior surface of the 16+ cage and the anion guest pair.17
The catalytic acceleration of this SNAr reaction in water was monitored optically by appearance of the strongly coloured DNP product at different H/DNFB concentrations. A routine kinetic analysis afforded a kcat/kuncat value of 23, with evidence to suggest that this catalysis is cavity-based: no such acceleration was observed with a smaller tetrahedral cage unable to accommodate DNFB but with a similar hydrophobic/cationic external surface. A Michaelis–Menten saturation experiment of reaction rate over a range of substrate concentrations yielded an apparent Km value of 7.3(±1.5) × 10−4 M, corresponding to a conventional binding constant of ≈ 1400 M−1 for DNFB: this could not be confirmed by a conventional titration experiment due to the catalysis happening when H and DNFB were combined.20
An interesting and unexpected observation was the occurrence of autocatalysis (i.e. catalysis by the DNP product) during the early stages of the reaction, as shown by a characteristic sigmoidal reaction profile at early times. We ascribed this to formation of a stacked DNFB/DNP pair in the cavity: the first traces of the DNP product formed facilitate binding of DNFB (present at that stage in large excess) in a hetero-guest pair, with one molecule of DNFB preferentially forming a H•(DNP)(DNFB) assembly in a cooperative manner rather than a simpler 1:1 H•DNFB complex. It is formation of this entity according to the equation H•(DNP) + DNFB = H•(DNP)(DNFB) that the Km value of 7.3(±1.5) × 10−4 M refers to. As the DNP product accumulates, for electrostatic reasons the two-guest complex H•(DNP)2 starts to dominate inside the 16+ cavity: the strong binding of this anionic guest pair dominates the speciation such that DNFB is unable to bind in the cage cavity after two equivalents of DNP have formed, at which point all of the cage cavity exists as H•(DNP)2, so is blocked to further substrate binding and the catalysis stops.
The ability of the cavity of H to accommodate a stacked pair of aromatic guests therefore has two apparently contradictory effects, as shown in Fig. 6. It accelerates the catalysis in the early stages by encouraging formation of the catalytically active mixed-guest species H•(DNP)(DNFB) in a cooperative manner, resulting in autocatalysis. DNFB binds more strongly to H•(DNP) than it does to empty H, because the remaining cavity space in singly-occupied H•(DNP) is perfectly complementary to a molecule of DNFB, not only in size/shape but also in the availability of an aromatic surface of DNP available to pi-stack with the second guest. However at higher product concentrations (after two complete reaction turnovers) the formation of more tightly-bound H•(DNP)2 inhibits the catalysed reaction. The opposed effects arising from the ability of the cavity to accommodate two guests [in the binding strength order H•(DNP)2 > H•(DNP)(DNFB) ≫ H•(DNFB)2, on electrostatic grounds] gives rise to some elaborate but interesting cage-mediated catalytic behaviour.20
 | ||
| Fig. 6 Progress of the SNAr reaction of DNFB (0.02–4.4 mM) with 0.01 M H as catalyst (pH 8.6, borate buffer) showing both autocatalysis at low substrate concentrations – note the characteristic sigmoidal shape of the reaction progress curve – and saturation of the cavity by product at high concentrations. | ||
4. Anion-binding to the M8L12 cage surface
Given the obvious importance of anion binding to the M8L12 cage surface that surfaced during the initial studies on cage-based catalysis, and which is a feature of all of the catalysis that we have observed with these cages, we wanted to study this in more detail. Most of the examples discussed are based on reaction of a guest with hydroxide ions, either in the cavity or around the exterior surface. However binding of other anion types also become apparent in our study on autocatalysis of the Kemp elimination: the reaction product (the 2-cyanophenolate anion) can accumulate around the cage surface, apparently binding more strongly than hydroxide does and therefore displacing it, due to the higher hydrophobicity of 2-cyanophenolate. As 2-cyanophenolate is itself a weak base it can deprotonate another cavity-bound benzisoxazole and thereby perpetuate the catalytic cycle (Fig. 7), leading to a situation where the reaction is accelerated by the product, giving the sigmoidal reaction profile characteristic of autocatalysis.21 | ||
| Fig. 7 Autocatalysis of the cage-catalysed Kemp elimination, with the product (2-cyanophenolate anion) displacing hydroxide from the cage portals and acting as the base to initiate another reaction cycle by deprotonation of cavity-bound 1,2-benzisoxazole. | ||
Another interesting example is the cage-catalysed aldol self-condensation of indane-1,3-dione.11 Formation of the product bindone necessarily requires an enolate anion to attack a neutral dione as the first step, so the presence of a catalysed reaction (which did not occur at all in the absence of Hw under the same conditions) implies that the enolate anions which will be present (given the modest pKa of ≈7 for indane-1,3-dione) accumulate around the cationic cage and are brought into close proximity with neutral diketone molecules whose association with the host is driven by the hydrophobic effect. Neutral indane-1,3-dione does bind inside the cavity, as shown by the crystal structure (Fig. 8, with the cavity containing a stacked pair of guests) and a solution NMR titration: and additional guests around the external surface (also shown in Fig. 8) show hydrogen-bonding interactions with the cage that imply that the external guests are actually the enolate anion reaction partners (though this is not completely clear from a crystal structure). Blocking the cavity with cycloundecanone does not stop the catalysis, implying that it occurs at the external surface: but the important point arising from these last two examples is that the cage surface can attract phenolate21 and enolate11 anions, and not just hydroxide ions, to participate in catalysed reactions.
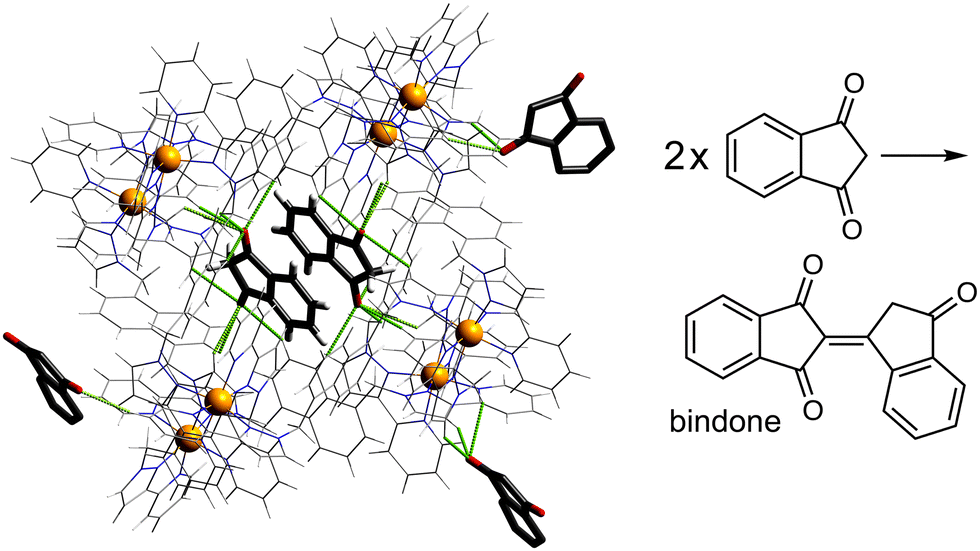 | ||
| Fig. 8 Cage-catalysed aldol condensation of indane-1,3-dione to bindone; the crystal structures show both cavity binding of neutral indane-1,3-dione guests (stacked pair across an inversion centre) plus external surface-binding of additional units presumed to be enolate anions, illustrating the ability of the cage to co-locate cavity-based (neutral) and surface-based (anionic) guests. | ||
An obvious question that arises is how to investigate and quantify the binding of anions to the cage surface, which is clearly desirable from the perspective of understanding and exploiting this behaviour further. One of the methods that we used to quantify binding of neutral molecules as guests inside the cage cavity is a fluorescence displacement assay. The M8L12 cage binds (and quenches the fluorescence of) 4-methyl-7-aminocoumarin (MAC) in water with a binding constant (2 × 104 M−1) that is readily determined from a simple fluorescence titration experiment.4b Stepwise addition of portions of a competing guest results in displacement of MAC from the cage cavity as the new guest competes for binding, restoring the fluorescence of MAC to an extent depending on the relative binding constants of MAC and the new guest, thereby allowing calculation of the value of K for the new guest.4b
We used a variant of this general method to investigate surface binding of anions to the M8L12 cage Hw.22 This required identification of an anionic fluorescent indicator which binds only to the cage surface (and not inside the cavity), and is quenched by the Co(II) ions when bound. Its fluorescence should be strong and in the visible region, away from the strong UV absorptions of the ligands in the cage; and finally binding needs to be strong enough to operate at the typical solution concentrations used for fluorescence measurements, but weak enough for the indicator to be displaced by accessible concentrations of competing anions.
Fluorescein (FLU) turned out to be ideal in all of these respects and is a di-anion in weakly basic conditions. Adding portions of cage Hw to fixed amount of fluorescein in buffered aqueous solution resulted in progressive quenching of the fluorescence as FLU bound to the cage surface; the resultant curve of fluorescence intensity vs. added cage concentration could be fitted to a 1:1 binding model giving K = 1.0 × 105 M−1 (Fig. 9a). Of course the cubic cage has six equivalent faces, so can in principle bind multiple FLU anions: but under the conditions of the experiment (excess of cage as the titration proceeds) both statistical and electrostatic considerations suggest that FLU anions will be dispersed amongst the cage surfaces rather than multiple FLU anions accumulating around one cage but not others, and 1:1 cage/FLU assembles will dominate. So this value of K = 1.0 × 105 M−1 can be taken as a 1:1 binding constant between Hw and FLU.
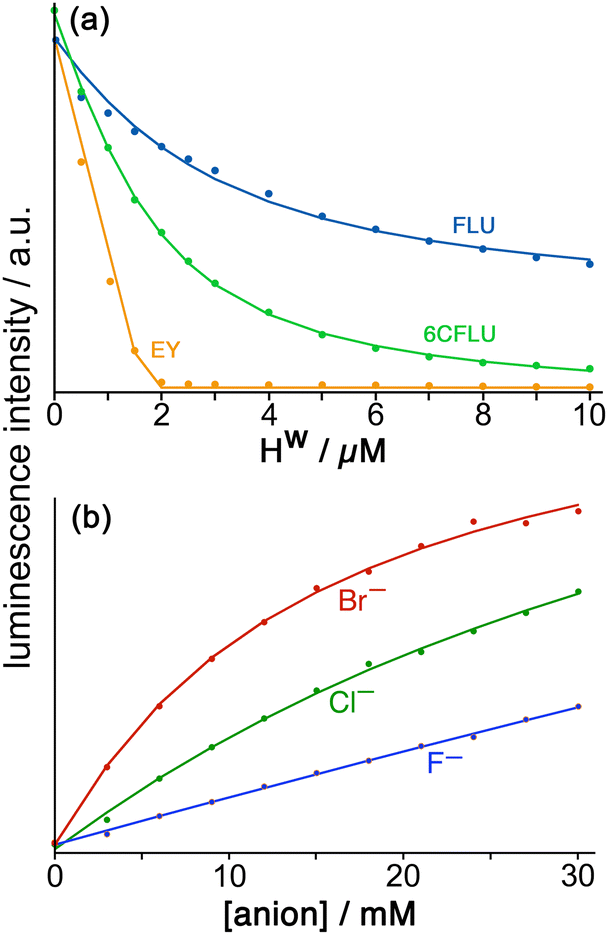 | ||
| Fig. 9 (a) Spectroscopic titrations of Hw with the fluorophores FLU (blue), 6CFLU (green) and EY (orange), each at 10 μM in water, showing in each case the progressive quenching of the fluorophore on addition of increasing amounts of Hw. The relative strengths of surface binding are clearly in the order FLU < 6CFLU < EY. (b) Results from the displacement assay showing how addition of portions of sodium salts with various anions to a solution containing Hw (50 μM) and FLU (5 μM) in water results in a steady increase in the fluorescence from FLU as it is displaced from the cage surface by the added anions which compete for the same sites, restoring the FLU emission. The measured data (small circles) could in all cases be fit to a 1:1 binding isotherm (solid lines) to give anion/cage binding constants. | ||
Interestingly a Job plot shows that when relative mole fractions of Hw and FLU are varied between 0:1 and 1:0, the maximum quenching of FLU occurs at a mole fraction of 0.83:0.17 FLU:Hw, indicative of formation of a 5:1 FLU:Hw assembly. This is strongly indicative of external binding of FLU at the windows in the cage surface, which would give a maximum of six equivalents of FLU binding to one Hw: evidently the sixth possible binding event – saturating the cage surface with FLU anions – is too weak to detect at the concentrations used. Note that there is no inconsistency with the observation of 1:1 FLU:Hw binding during the titration, as that was done with excess Hw relative to FLU by the end of the titration, whereas observation of the 5:1 FLU:Hw assembly requires a large excess of FLU relative to Hw. Again we see how experiments done under different conditions need to be combined to give a clearer picture of the host/guest behaviour of this cage.19
Stepwise addition of a range of different anions to a FLU/Hw combination in aqueous solution resulted in progressive restoration of fluorescence, as FLU was displaced from the cage surface by increasing amounts of the competing anion (Fig. 9b). The resultant increases in fluorescence with anion concentration could be fit to 1:1 binding isotherms using the standard methodology for a displacement assay, taking account of the known binding constant of FLU. The resulting numerical binding constants effectively assume that each binding site of the cage (one face) is independent of all the others. The key point here is the differences between anion types which show that (i) for a wide range of monoanions the affinity for the cage follows ease of desolvation in water, which is the basis of the Hofmeister series (e.g. F− < Cl− < Br−); (ii) addition of hydrophobic alkyl chains to anions makes little difference (e.g. acetate, propionate, butyrate all show similar binding to Hw) which implies that those alkyl groups remain projecting into the aqueous solvent when the carboxylate terminus binds, such that their environment does not significantly change; and (iii) dianions bind more strongly than monoanions for obvious electrostatic reasons.22
In addition to this quantification of binding of small anions to Hw we evaluated other large fluorescent aromatic anions beyond FLU for binding to Hw in water (Fig. 9a), and found even stronger binding with 6-carboxyfluorescein (6CFLU: K ≈ 4 × 105 M−1), presumably due to its 3– charge; with Eosin-Y (EY) which is more hydrophobic than FLU because of its Br atom substituents; and hydroxypyrene-tris-sulfonate, with these last two having binding that is too strong to extract a binding constant under the conditions used (hence K > 106 M−1 for both). In all cases Job plots at different mole ratios showed maximal quenching of these anionic, aromatic fluorophores at Hw:fluorophore ratios of between 1:4 and 1:6, consistent with external-surface binding to the cage faces.22
We could also indirectly evaluate anion binding affinities to the cage surface by their effect on the rate of a catalysed reaction. Replacing the substrate 1,2-benzisoxazole by 5-nitro-1,2-benzisoxazole results in cage-based catalysis of the Kemp elimination reaction with hydroxide which occurs (as described earlier) at the cage external surface, the product in this case being 2-cyano-4-nitrophenolate.9 The accumulation of anions around the cage surface can slow down the catalysed Kemp elimination reaction of 5-nitro-1,2-benzisoxazole simply by competitive displacement of the hydroxide ions from the cage surface, close to the substrate. Addition of a fixed amount of different anions therefore retards the catalysed reaction to different extents, depending on the anion/cage surface affinity, and the rate of the catalysed Kemp elimination of 5-nitro-1,2-benzisoxazole is therefore an easily-measurable proxy for the binding affinity of the different anions to the cage. The results9 are consistent with the affinity order that was apparent using the fluorescence displacement assay described above,22 with retardation of the reaction following the Hofmeister series for many anions indicating that their affinity for the cage surface follows their ease of desolvation, with the more hydrophobic anions (nitrate, bromide) causing a reduction in the rate constant for the 5-nitro-1,2-benzisoxazole/HO− reaction around the cage surface by ca. 1 order of magnitude. Interestingly anions that are weakly basic (e.g. HCO3−, acetate, and even fluoride) accelerate the reaction slightly: although they will necessarily displace HO− from around the cage, they can take its place by deprotonating the substrate to initiate the reaction,9cf. the participation of 2-cyanophenolate as base in the autocatalyzed Kemp elimination reaction with unsubstituted benzisoxazole.21 Thus, useful mechanistic insights concerning cage/anion interactions can be obtained from their effect on catalysis.
An important and potentially general feature of this catalysis is that the interactions that bring together the two reaction partners are orthogonal, implying that any of a wide range of surface-bound anions can be brought into close proximity with any of a wide-range of neutral, and usually cavity-bound, guests. The orthogonality of the two types of binding was shown in the solid state by crystalline sponge experiments, and in solution by fluorescence displacement of one of two different types of guest.23 A standard crystalline sponge experiment, soaking crystals of H in a concentrated MeOH solution containing both a cavity binding guest (MAC) and a surface-binding ion (iodide, as its Bu4N+ salt) in MeOH, resulted in uptake of both guest types into their different positions: MAC in the cavity and iodide surrounding and outside the cage, in particular at the six ‘portals’ in the cage faces surrounding the guest. Use of a range of other surface-binding ions (nitrate, sulfate, hexafluorophosphate) in conjunction with MAC gave exactly similar results with both neutral and anionic guests being taken up into their distinct sites inside or around the cavity of H (Fig. 10). Thus, two different guest types can be taken up into a host crystalline array in a single step because the different guest types go to different sites.23
 | ||
| Fig. 10 Results of crystalline sponge experiments using H as the host in which a cavity-bound guest (4-methyl-7-aminocoumarin, MAC) and surface bound guest anions are taken up to different sites in the same experiment. In (a) and (b) the surface-bound anion is iodide; in (c) and (d) it is sulfate. The wireframe cage views in (a) and (c) emphasise the location of the MAC guest; the space-filling views in (b) and (d) emphasise the locations of the anions in the surface portals. | ||
In solution, the orthogonality of the two types of binding can be shown by selective displacement from Hw of either surface-bound or cavity-bound fluorophores according to the type of guest, and these generate different fluorescence responses (Fig. 11).23 In the presence of both FLU and MAC in aqueous solution, the large and anionic FLU associates with the cage exterior surface, as described earlier,22 and its green fluorescence is quenched: the smaller, neutral MAC binds inside the cage cavity, and in doing so its blue fluorescence is likewise quenched.4b A competing guest will displace one or other of these according to whether the guest is a surface-binder or a cavity-binder, and this is signalled by the colour of the fluorescence change as one fluorophore has its fluorescence restored.
 | ||
| Fig. 11 (a) Cartoon representation of the dual fluorescence displacement assay used to probe cavity vs. surface binding of guest types in host Hw. (b) A CIE diagram showing the fluorescence colour changes associated with addition of (surface-binding) ascorbate, (cavity-binding) cyclooctanone, or chloride, to a mixture of Hw (150 μM), MAC (10 μM), FLU (20 μM) and Ru (30 μM). The two sets of dashed arrows starting at different starting points indicate the directions of colour shifts generated by different analytes using 365 nm (black arrows) or 395 nm excitation (blue arrows), respectively. These arrows are just to aid the eye: the end-points of each titration are illustrated by the coloured points. | ||
Thus, addition of ascorbate to an Hw/FLU/MAC mixture in aqueous solution results in an increase only in green fluorescence as the FLU is displaced from the cage surface; addition of cyclooctanone to the same mixture results in an increase only in blue fluorescence as the MAC is displaced from the cage cavity. This provides a straightforward colorimetric indication of where a guest binds. Intriguingly in one case (chloride) we saw an intermediate luminescence colour change with increases in both blue and green components (Fig. 11),23 suggesting that chloride is not just a surface-binder but can also bind inside the cage cavity. This helps to explain why Hw as its chloride salt gives smaller guest binding constants for many guests than it does as its BF4− salt – because chloride can compete to occupy the cavity, though all crystal structures only show it at the surface sites.21
Overall, the ability to bring any of a wide range of anions (around the cage surface) into close proximity with and surrounding and of a wide range of neutral hydrophobic reaction partners (in the cavity) – generating a high local concentration of the anions surrounding the neutral guest – has obvious scope to be the basis of a wide range of cage-catalysed reactions.
5. Catalysis of oxidation reactions based on cage redox activity
In this final section we summarise recent progress in a quite different area: it is related to the general principles underlying the cage-based catalysis that we have discussed above (particularly, the co-location of neutral and anionic reaction partners using orthogonal interactions) but operates in a quite distinct way by exploiting the cage-based redox activity associated with Co(II)/Co(III) couples as a way to initiate the reaction.H2O2 and the peroxymonosulfate anion (PMS; HSO5−) are well-known strong oxidants with very wide synthetic utility; both have been of interest in ‘advanced oxidation processes’ for wastewater purification in a clean way that produces just water as a byproduct.24,25 A feature of both however is that on their own they react slowly with substrates and therefore have limitations to their effectiveness for kinetic reasons, despite their high positive redox potentials. In both cases they can be activated by a redox reaction with an appropriate oxidisable metal ion, with Co2+ being a typical example, which generates highly reactive radical species that oxidise substrates far more quickly.
For H2O2 the relevant reactions24a (by analogy with the well-known Fenton chemistry based on Fe3+/H2O2) are plausibly
| Co2+ + H2O2 = Co3+ + HO˙ + HO− |
| Co3+ + H2O2 = Co2+ + HOO˙ + H+ |
| Co2+ + HSO5− = Co3+ + SO4˙− + HO− |
In both cases the reactive radicals can be generated by reaction of the primary oxidant (H2O2 or PMS) with Co(II) ions in the cage superstructure, generating the reactive radicals around the cage surface immediately surrounding any cavity-bound guests, which is the basis of the catalysis examples shown below. In addition the anionic radical SO4˙− will be attracted to the cage surface for the electrostatic reasons discussed earlier, further increasing its tendency to accumulate surrounding any cavity-bound guests.
The first example of this type of metal-catalysed oxidation of a cavity-bound guest is the oxidation of luminol by H2O2 using the cubic cage Hw (Fig. 12).26 Oxidation of luminol using H2O2 is accompanied by chemiluminescence – which is the basis of its use as a forensic analytical tool in e.g. detection of bloodstains. Luminol is the right shape and size to be a good guest for the cage cavity [K = 1.26(6) × 104 M−1], and addition of H2O2 an Hw/luminol mixture in aqueous borate buffer at pH 8.5 generates chemiluminescence arising from oxidation of the cavity-bound guest.
 | ||
| Fig. 12 Cartoon representation of the oxidation of luminol by H2O2 catalysed by the cage Hw which (i) binds the guest, (ii) uses its Co(II)/Co(III) redox couple to activate the H2O2 generating reactive oxygen species surrounding the cage surface, and (iii) tethers any hydroxide ions required for the reaction in place via the cage positive charge. | ||
Appropriate control experiments confirm the two essential points that (i) catalysis by the cage requires the Co(II)/Co(III) couple, and (ii) the luminol needs to be cage-bound to be oxidised (proximity of reaction partners). The first control experiment was to replace the Co(II) ions in the cage Hw by Zn(II) ions, which generates an isostructural M8L12 cage but without the redox activity needed to activate the H2O2: this resulted in the complete loss of any catalysed luminol oxidation by H2O2. The second control experiment was to have an equivalent number of Co(II) centres present, but as separated mononuclear complexes (i.e. 8 mononuclear complexes rather than one Co8 cage) such that no cavity-binding of luminol could occur. Whilst the same activation of H2O2 by the Co(II) centres would be present, in this case the reactive oxidants are not being generated so as to surround a cavity-bound guest: the Co8 cage has, effectively, been disassembled. This resulted in almost complete loss of catalytic activity. This pair of control experiments demonstrates that catalysis of the oxidation of luminol by H2O2 requires both that the luminol is cavity-bound in a cage, and that the cage contains Co(II)/Co(III) couples which allows the reactive oxygen species to be generated at the cage surface close to, and surrounding, the guest.26 Importantly this final oxidation step can also involve hydroxide ions – which are already anchored in place by the cationic cage surface, so are ready and waiting.24a
A conceptually similar example is provided by oxidation of fluorescein using PMS, catalysed by the larger cuboctahedral cage denoted Co12.27 This cage (Fig. 13)28 has a cavity volume around 2.5× larger than that of Hw, which is large enough to bind strongly one molecule of fluorescein. In the absence of a crystal structure we cannot prove that the guest binds inside the cavity compared to binding at the exterior surface, but the strength of the binding (logK = 6.7) and the observation of a 1:1 Co12:fluorescein ratio for the host/guest assembly are together strongly suggestive of this.28
 | ||
| Fig. 13 Three representations of the structure of the cuboctahedral cage Co12. (a) A view illustrating the binding modes of the distinct tritopic (red) and ditopic (blue) ligands; (b) a cartoon sketch emphasising the combination of tritopic (red) and ditopic (grey) ligands around the Co12 superstructure; (c) a crystal structure of one of the three diastereoisomers of the complex cation. | ||
On addition of PMS to a Co12/fluorescein mixture in aqueous solution (with some surfactant present to help solubilise the Co12 cage in water), fluorescein is quickly destroyed by oxidation (97% in 20 minutes), as shown by the loss of its characteristic UV/vis absorbance (Fig. 14).28 As with the luminol/Hw example mentioned above,26 two control experiments were carried out. Firstly, use of the isostructural Zn12 cage prevented any degradation of fluorescein by PMS, confirming that the ability of PMS to oxidise fluorescein relies on activation of PMS to SO4˙− by the Co(II) ions in the cage superstructure. Co-location of fluorescein and PMS in and around the M12 cage cavity alone is an insufficient condition for catalysis: the redox-based activation of PMS to generate SO4˙− is also required. Secondly, ‘disassembling’ the cage by having an equivalent number of Co(II) ions in the form of mononuclear complexes removes most of the catalytic effect, confirming the importance of the co-location of fluorescein (in the cavity) and SO4˙− ions (around the cage surface) which the intact cage provides. Other dyes such as rhodamine and methylene blue were likewise efficiently destroyed by oxidative degradation using PMS, catalysed by the Co12 cage.28
 | ||
| Fig. 14 Cartoon representation of the oxidative degradation of fluorescein by peroxymonosulfate, catalysed by the cage Co12 which again performs three functions: (i) it binds the fluorescein; (ii) it provides the necessary redox activation of peroxymonosulfate to generate the reaction SO4˙− radical anion; and (iii) its positive charge attracts the SO4˙− radical anions to the cage surface, resulting in accumulation around the fluorescein substrate. | ||
In these two recent cases the cages act not just to bind the substrate and accumulate the reaction partners around the surface, but also to provide redox-based activation of the oxidising agent as the basis of the catalysis. This type of direct participation of redox-active ions in the cage superstructure in catalysed reactions, such that the cage is no longer just acting as a charged but hydrophobic reaction vessel, offers substantial promise for use of the cages in redox- and photo-redox catalysis processes, and this is the focus of ongoing work. Indeed the redox participation of cage components is central to work summarised in recently reviews work by Duan and co-workers,1r and Reek and co-workers,1s on the light-induced reactions of guests bound in cage/guest assemblies which similarly exploit the combination of redox activity and preorganisation that cages can provide.
6. Conclusions and future directions
The recent results summarised in this review provide significant advances to our understanding of cage-based catalysis, and suggest multiple new opportunities. The main conclusions may be summarised as follows:(i) The catalytic effect relies on proximity of neutral substrates which interact with the cage via the hydrophobic effect, and the anionic reaction partners which accumulate around the cage due to electrostatic effects; these two types of cage/guest interaction can be considered as orthogonal and provide a simple means to allow accumulation of a high local concentration of anions around a potentially reactive substrate. This presents a potentially very general means to effect cage-based catalysis of organic molecules with anions.
(ii) It's not all about the cavity! Whilst the most effective catalysis appears to occur with cavity-bound substrates, association of a hydrophobic substrate with the cage exterior surface still brings it into proximity with the ‘shell’ of anionic reactions partners (often, but not necessarily, hydroxide), allowing catalysis to occur for guests that do not bind well inside the cage cavity.
(iii) The relative binding strengths of anions to the cage surface could be investigated quantitatively, by both a fluorescence displacement assay, and by the inhibiting or accelerating effect of different anions on the rates of a catalysed reaction, revealing some interesting patterns such as correlation of binding strengths of mono-anions with the Hofmeister series.
(iv) The redox activity of the Co(II) ions which are often used to prepare our cage family provides an entirely new type of cage-based catalysis using ‘advanced oxidation process’ methodology: viz. cavity-bound substrates can undergo catalysed oxidation by H2O2 or by peroxymonosulfate following activation of these oxidants via a redox reaction with the cage superstructure, opening the door to new types of redox-based catalysis in which the cage plays an active role beyond binding and co-locating the reaction partners.
All of this points to some interesting possible future directions, with two being worth highlighting. Firstly: we, like many other groups, have been able to identify examples of cage-based catalysis: with the underlying mechanism being the ability of the host cage to bind multiple different guests as reaction partners at different sites (cavity and surface). The importance of this has been highlighted recently in reviews by both Nitschke1t and Lusby1q who have emphasised the importance of ‘dual binding’ as a basis for some types of catalysis (in contrast to, for example ‘constrictive binding’ where a single bound guest has its reactivity changed). This substantially complicates the traditional view of a cage as a box that binds a single type of guest – a view that remains implicit in the language of ‘host–guest’ chemistry. Indeed the role of cage in bringing together multiple reaction components might be considered more like the role of a surface in heterogenous catalysis at bringing together and activating >1 reactant type.
Secondly: the participation of metal ions from the cage superstructure in redox-based catalysis (Section 5, above) provides an additional dimension to the complexity of what may be possible, with the host/guest (or, following the above comments, host/multiple guests) properties of the cage being the starting point for a new level of sophistication in catalysis using supramolecular assemblies. The ultimate expression of this in e.g. artificial photosynthesis remains a distant goal, but the field has reached the stage where all of the necessary component capabilities (multiple guest binding; activation of substrates; incorporation of arrays of chromophores in a spatially well-defined array; redox activity; and even multiple sequential reactions) have been individually demonstrated.1 So future developments, arguably, will involve bringing these together into single catalytic systems.
Data availability
As this is a review article summarising already-published work, and containing no new results, no data availability statement is necessary.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to thank very much all of the talented researchers in my group who contributed to this work; their names are listed in the references. Particular thanks are due to my collaborators Dr Genevieve Dennison (Australian Defence Science Technology Group), who managed the work with chemical warfare agents in ref. 12; and Assoc. Prof. Kellie Tuck (Monash University) whose research group did all other experimental work associated with ref. 12 and 20 based on samples of cages provided by my group at Warwick. Thanks are also due to the various funding agencies who have supported this work and the postdocs/PhD students who did it: EPSRC, The Leverhulme Trust, The Royal Society, the European Union, the universities of Warwick and Monash, the Swiss National Science Foundation, the government of Thailand, and the UK Diamond Light Source. Specific funding details are given in the individual papers in the reference list.Notes and references
- (a) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed; (b) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS; (c) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (d) D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423 CrossRef CAS PubMed; (e) H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171 CrossRef CAS; (f) E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480 CrossRef PubMed; (g) F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204 CrossRef; (h) D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563 CrossRef CAS; (i) Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707 RSC; (j) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed; (k) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS; (l) M. Otte, ACS Catal., 2016, 6, 6491 CrossRef CAS; (m) C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447 CrossRef CAS; (n) W.-X. Gao, H.-N. Zhang and G.-X. Jin, Coord. Chem. Rev., 2019, 386, 69 CrossRef CAS; (o) L. Zhao, X. Jing, X. Li, X. Guo, L. Zeng, C. He and C. Duan, Coord. Chem. Rev., 2019, 378, 151 CrossRef CAS; (p) M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969 CrossRef CAS; (q) T. Piskorz, V. Martí-Centelles, R. L. Spicer, F. Duarte and P. J. Lusby, Chem. Sci., 2023, 14, 11300 RSC; (r) X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100 CrossRef CAS; (s) R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Chem. Rev., 2023, 123, 5225 CrossRef CAS; (t) F. J. Rizzutto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204 CrossRef.
- M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073 CrossRef CAS.
- I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS.
- (a) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744 RSC; (b) S. Turega, W. Cullen, M. Whitehead, C. A. Hunter and M. D. Ward, J. Am. Chem. Soc., 2014, 136, 8475 CrossRef CAS.
- W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 2790 RSC.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231 CrossRef CAS PubMed.
- C. G. P. Taylor, A. J. Metherell, S. P. Argent, F. M. Ashour, N. H. Williams and M. D. Ward, Chem. – Eur. J., 2020, 26, 3065 CrossRef CAS PubMed.
- A. B. Solea, B. Sudittapong, C. G. P. Taylor and M. D. Ward, Dalton Trans., 2022, 51, 11277 RSC.
- M. D. Ludden, C. G. P. Taylor, M. B. Tipping, J. S. Train, N. H. Williams, J. C. Dorratt, K. L. Tuck and M. D. Ward, Chem. Sci., 2021, 12, 14781 RSC.
- C. Mozaceanu, C. G. P. Taylor, J. R. Piper, S. P. Argent and M. D. Ward, Chemistry, 2020, 2, 22 CrossRef CAS.
- B. Sudittapong, C. G. P. Taylor, J. Williams, R. J. Griffiths, J. R. Hiscock and M. D. Ward, RSC Adv., 2024, 14, 26032 RSC.
- J. C. Dorratt, R. J. Young, C. G. P. Taylor, M. B. Tipping, A. J. Blok, D. R. Turner, A. I. McKay, S. Ovenden, M. D. Ward, G. H. Dennison and K. L. Tuck, Dalton Trans., 2023, 52, 11802 RSC.
- C. G. P. Taylor, J. R. Piper and M. D. Ward, Chem. Commun., 2016, 52, 6225 RSC.
- (a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, S. Matsunaga, K. Takada, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 CrossRef CAS PubMed; (b) M. Hoshino, A. Khutia, H. Xing, Y. Inokuma and M. Fujita, IUCrJ, 2016, 3, 139 CrossRef CAS PubMed.
- N. T. Johnson, P. G. Waddell, W. Clegg and M. R. Probert, Crystals, 2017, 7, 360 CrossRef.
- (a) C. G. P. Taylor, S. P. Argent, M. D. Ludden, J. R. Piper, C. Mozaceanu, S. A. Barnett and M. D. Ward, Chem. – Eur. J., 2020, 26, 3054 CrossRef CAS PubMed; (b) C. G. P. Taylor, J. S. Train and M. D. Ward, Chemistry, 2020, 2, 510 CrossRef CAS.
- A. J. Metherell and M. D. Ward, Dalton Trans., 2016, 45, 16096 RSC.
- (a) M. R. Ams, D. Ajami, S. L. Craig, J. S. Yang and J. Rebek, J. Am. Chem. Soc., 2009, 131, 13190 CrossRef CAS PubMed; (b) S. Mecozzi and J. Rebek, Chem. – Eur. J., 1998, 4, 1016 CrossRef CAS; (c) J. Rebek, Acc. Chem. Res., 2009, 42, 1660 CrossRef CAS.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC; (b) F. Ulatowski, K. Dabrowa, T. Balakier and J. Jurkzac, J. Org. Chem., 2016, 81, 1746 CrossRef CAS; (c) D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792 RSC.
- J. C. Doratt, C. G. P. Taylor, R. J. Young, A. B. Solea, D. R. Turner, G. H. Dennison, M. D. Ward and K. L. Tuck, Chem. – Eur. J., 2024, 30, e202400501 CrossRef.
- W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, J. Am. Chem. Soc., 2018, 140, 2821 CrossRef CAS PubMed.
- M. D. Ludden and M. D. Ward, Dalton Trans., 2021, 50, 2782 RSC.
- M. D. Ludden, C. G. P. Taylor and M. D. Ward, Chem. Sci., 2021, 12, 12640 RSC.
- (a) A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121 CrossRef CAS; (b) P. Kumari and A. Kumar, Res. Surf. Interfaces, 2023, 11, 100122 CrossRef; (c) D. Ma, H. Yi, C. Lai, X. Liu, X. Huo, Z. An, L. Li, Y. Fu, B. Li, M. Zhang, L. Qin, S. Liu and L. Yang, Chemosphere, 2021, 275, 130104 CrossRef CAS PubMed.
- (a) J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502 CrossRef CAS; (b) F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41 CrossRef CAS; (c) X. Zheng, X. Niu, D. Zhang, M. Lv, X. Ye, J. Ma, Z. Lin and M. Fu, Chem. Eng. J., 2022, 429, 132323 CrossRef CAS; (d) J. Lee, U. von Gunten and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 3064 CrossRef CAS PubMed.
- A. B. Solea and M. D. Ward, Dalton Trans., 2023, 52, 4456 RSC.
- S. P. Argent, F. C. Jackson, H. M. Chan, S. Meyrick, C. G. P. Taylor, T. K. Ronson, J. P. Rourke and M. D. Ward, Chem. Sci., 2020, 11, 10167 RSC.
- X. Zhang, B. Sudittapong and M. D. Ward, Inorg. Chem. Front., 2023, 10, 1270 RSC.
| This journal is © The Royal Society of Chemistry 2024 |