DOI:
10.1039/D4CC03431G
(Feature Article)
Chem. Commun., 2024,
60, 9007-9021
Advances in electrocatalytic dehydrogenation of ethylamine to acetonitrile
Received
9th July 2024
, Accepted 29th July 2024
First published on 29th July 2024
Abstract
The electrocatalytic dehydrogenation of ethylamine (EDH), owing to its high hydrogen content, holds broad prospects in electrochemical hydrogen (H2) production, H2 storage, and addressing energy issues, thus deserving wide attention. In this feature article, we first summarized the fundamentals of thermocatalytic and electrocatalytic EDH and reviewed the recent state-of-the-art advances in catalyst research, specifically platinum group metal (PGM) catalysts and non-PGM catalysts. We systematically discussed the potential applications of electrocatalytic EDH in energy storage and conversion. Finally, we provide our perspective on the key challenges and future developments in this field. We believe this feature article will offer helpful guidance for oriented design and optimization of stable and efficient catalysts for electrocatalytic EDH and related energy applications.
1. Introduction
Ethylamine and acetonitrile are two important chemical raw materials that play crucial roles in modern chemical and pharmaceutical industries.1–4 Ethylamine, serving as an intermediate for the synthesis of many organic compounds, finds wide applications in the production processes of pharmaceuticals, insecticides, and rubber chemicals.5–7 On the other hand, acetonitrile is commonly used as a solvent in chemical reactions, particularly vital in the pharmaceutical industry for synthesizing peptides and other drug compounds.8–10 Additionally, it plays a role in plastic and fiber manufacturing and serves as a solvent for lithium batteries.11–13 In recent years, with the growing awareness of environmental protection and the pursuit of renewable energy, electrocatalytic synthesis technology has gradually gained attention as an environmentally friendly approach.14–18 The direct dehydrogenation of ethylamine (EDH) to acetonitrile and the direct hydrogenation of acetonitrile (AHD) to ethylamine are considered feasible processes, and electrocatalytic synthesis technology provides a green and efficient pathway for the preparation of these two compounds. Notably, with the transition from traditional fossil fuels to clean energy, the concept of liquid organic hydrogen carriers (LOHC) is receiving increasing attention.19,20 In this context, technologies such as electrocatalytic EDH and electrocatalytic AHD have been found to have broad applications in hydrogen (H2) storage and energy conversion.21,22 The development of these technologies not only helps improve energy utilization efficiency but also holds the promise of advancing the development and application of clean energy, contributing to future sustainable development efforts.
H2 is hailed as a clean and promising alternative to traditional fossil fuels, holding crucial significance in alleviating global energy shortages and environmental pollution.23,24 Utilizing renewable energy to produce H2 through water electrolysis is now widely regarded as one of the most promising green energy options.25 Unfortunately, the realization of H2 economy is hindered by challenges associated with efficient H2 storage and transportation due to its low volumetric density under ambient conditions.19,26,27 So far, H2 is typically stored either as a compressed gas or in liquid form, both of which often require extremely high pressure or low temperatures. Additionally, these storage methods suffer from low energy efficiency and fail to meet the requirements for H2 distribution and volumetric density during transport.28 Currently, finding more efficient, cost-effective, high-density, and convenient methods for H2 storage remains challenge. Inspired by these issues, an alternative option for H2 storage and transportation with the aforementioned advantages is the use of LOHC systems, which has sparked widespread research interest. For long-distance transportation of large amounts of H2, LOHC presents an up-and-coming alternative. Moreover, LOHC can leverage existing infrastructure such as oil and natural gas pipelines for storage and transportation, not only saving costs but also facilitating large-scale practical applications.29 However, it is necessary to develop LOHC compounds that meet various standards, including thermal stability, chemical stability, energy efficiency, low toxicity, sustainability, and high H2 storage capacity.30 To address this issue, researchers have explored various LOHCs for H2 uptake and release. Among these LOHC carriers, ethylamine exhibits significant potential for electrochemical H2 storage due to its high theoretical H2 content (gravimetric 8.9 wt% H2; volumetric 72.1 gH2 L−1), low redox potential (0.13 V vs. RHE),22 and intriguing redox activity at ambient conditions. Ethylamine can be obtained through processes such as ethanol amination,31,32 reductive ammoniation of acetaldehyde33,34 and AHD,35,36 ensuring its abundance and accessibility. EDH and AHD on aqueous phase over electrocatalysts is a promising method for H2 release and uptake as it operates at ambient temperature and demonstrates high recyclability.
However, practical applications require highly active, selective, and stable electrocatalysts. In the past few years, considerable reports on electrocatalytic AHD have emerged,36–43 mainly based on Cu-based catalysts to suppress the occurrence of the H2 evolution reaction (HER) and to enhance product selectivity. Additionally, the exothermic AHD is thermodynamically favorable, which can be easily achieved using traditional catalysis in ambient conditions. Therefore, the principal obstacle in this research lies in the thermodynamically unfavorable EDH, a topic that remains relatively novel with sporadic reports over the past few years, predominantly relying on platinum group metal (PGM) catalysts. However, the high cost and limited reserves of PGMs hinder the widespread application of EDH. Furthermore, there is an insufficient fundamental understanding of the electrocatalysis mechanism and catalyst deactivation for this reaction. Thus, now is an opportune time to provide a comprehensive summary of EDH. This feature article summarizes the latest research progress on electrocatalytic EDH and explores its potential applications in H2 storage, regenerative fuel cells and batteries (Scheme 1). By examining the EDH electrocatalysis mechanism, the catalyst design principles, and potential application prospects, we hope to promote further research in this field and speed up the development of the H2 economy. We also believe that this feature article will advance the development of novel electrocatalysts for EDH and their potential applications and deepen the understanding of the inherent mechanism of electrocatalytic EDH.
 |
| | Scheme 1 A brief description of the whole content of this feature article toward the dehydrogenation of ethylamine (EDH) to acetonitrile. | |
2. Fundamentals of catalytic and electrocatalytic ethylamine dehydrogenation
2.1 Physical and chemical properties of ethylamine
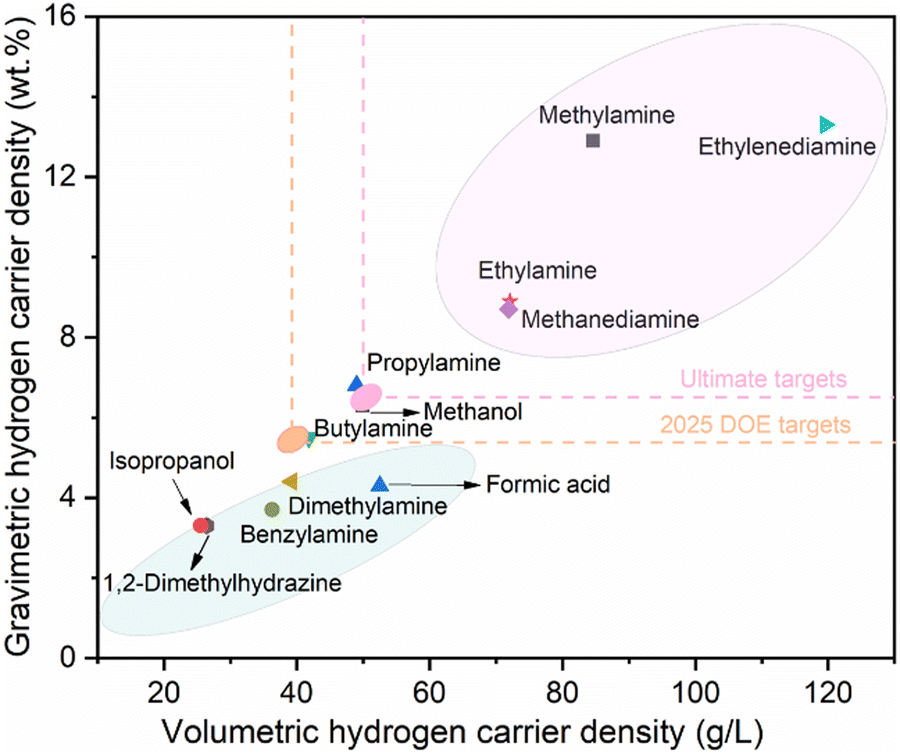
Ethylamine is an organic compound with the chemical formula C2H7N, one of the simplest aliphatic amines. It is a colorless gas at standard temperature and pressure, possessing a strong ammonia-like odor, and is highly soluble in water, ethanol, ether, and other solvents. Various characteristic properties of common LOHC compounds, including typical amines, alcohols and carboxylic acid are summarized in Table 1, encompassing their dehydrogenation product, boiling point, redox potential, mass hydrogen carrier density and volume carrier density. To provide a clear and more intuitive comparison of the theoretical hydrogen energy carrier density, Fig. 1 displays the hydrogen carrier density of these common LOHC compounds. It is clear that except for methylamine and ethylenediamine, ethylamine exhibits the highest mass and volume hydrogen carrier density, which is a significant indicator for assessing the feasibility of large-scale practical applications, and we will discuss this in detail in the application section. More importantly, compared to the dehydrogenation product of ethylamine, acetonitrile, the dehydrogenation product of methylamine, hydrogen cyanide, and the dehydrogenation product of ethylenediamine, cyanogen, exhibit higher toxicity.
Table 1 Important physiochemical properties of common liquid organic hydrogen carrier (LOHC) compounds
| LOHC compounds |
Dehydrogenation product |
Boiling point of LOHC/product (°C) |
Redox potential (V vs. RHE) |
Gravimetric hydrogen carrier density (wt%) |
Volumetric hydrogen carrier density (g L−1) |
| CH3OH methanol |
HCHO Formaldehyde |
65/−19 |
0.33 |
6.3 |
49.9 |
| CH3CHOHCH3 isopropanol |
CH3C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OCH3 Acetone OCH3 Acetone |
83/56 |
0.12 |
3.3 |
25.9 |
| HCOOH formic acid |
CO2 Carbon dioxide |
101/−78.5 |
−0.17 |
4.3 |
52.5 |
| CH3NH2 methylamine |
HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N Hydrogen cyanide N Hydrogen cyanide |
−6/26 |
0.24 |
12.9 |
84.6 |
| CH3CH2NH2 ethylamine |
CH3CN Acetonitrile |
17/81 |
0.13 |
8.9 |
72.1 |
| CH3(CH2)2NH2 propylamine |
CH3CH2CN Propionitrile |
48/97 |
0.15 |
6.8 |
49.0 |
| CH3(CH2)3NH2 butylamine |
CH3 (CH2)2CN Butyronitrile |
78/118 |
0.15 |
5.5 |
41.8 |
| NH2CH2NH2 methanediamine |
NC–NH2 Cyanamide |
118/260 |
∼0.20 |
8.7 |
71.9 |
| CH3NHCH3 dimethylamine |
H2CN–CH3 Schiff base |
8/251 |
∼0.20 |
4.4 |
39.2 |
| NH2(CH2)2NH2 ethylenediamine |
NC–CN Cyanogen |
116/−21 |
∼0.20 |
13.3 |
119.6 |
| CH3NHNHCH3 1,2-dimethylhydrazine |
H3C–NN–CH3 Azomethane |
87/95 |
∼0.20 |
3.3 |
26.4 |
| C6H5CH2NH2 benzylamine |
C6H5CN Benzonitrile |
184/190 |
0.19 |
3.7 |
36.3 |
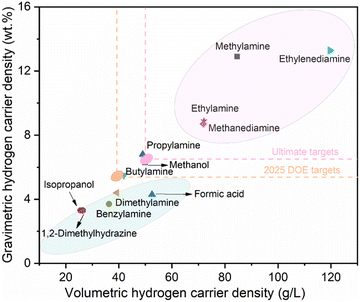 |
| | Fig. 1 Comparison of hydrogen carrier density of common liquid organic hydrogen carrier (LOHC) compounds. | |
2.2 Reaction thermodynamics
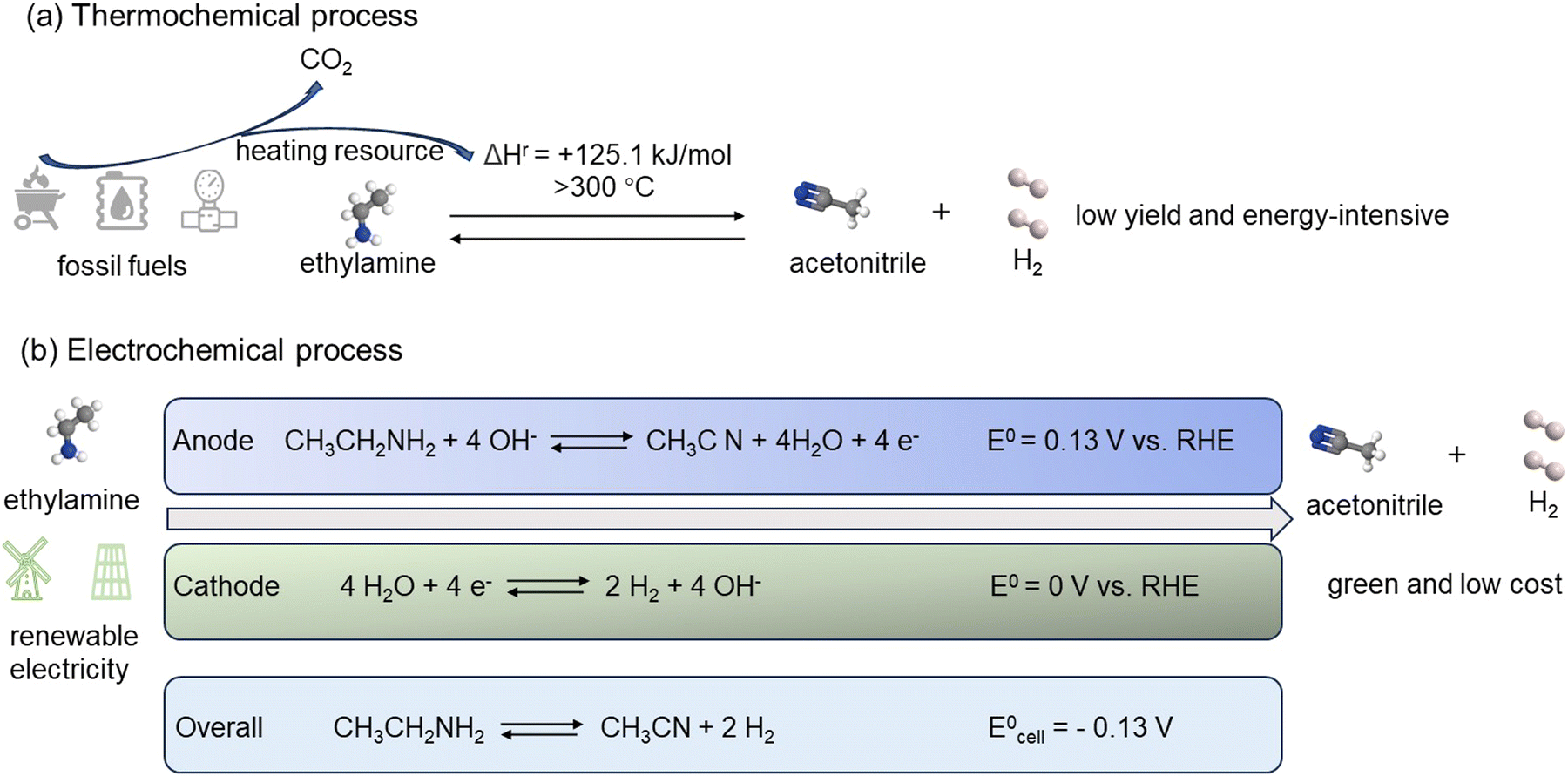
Like other typical dehydrogenation reactions, EDH is also an endothermic reaction, with a reaction enthalpy change of +125.1 kJ mol−1. This positive reaction enthalpy change makes the conversion of ethylamine into acetonitrile and H2 highly unfavorable thermodynamically. The Gibbs free energy of EDH to form acetonitrile and H2 under standard conditions can also be calculated from the formula ΔGr = ΔHr − TΔSr = +58.4 kJ mol−1. This corresponds to a tiny equilibrium constant on the order of 10−11. For endothermic reactions, the equilibrium constant increases with increasing temperature. Therefore, to achieve an equilibrium constant of 1, a temperature of nearly 300 °C or even higher is required to approach a significant conversion. Such high temperatures often are not desired for applications and require the combustion of fossil fuels to provide heat. Meanwhile, this process also generates CO2, which is detrimental to the environment (Fig. 2(a)). Researchers have investigated whether dehydrogenation of amines can be achieved at moderate temperatures (110–130 °C).44 At these temperatures, the thermodynamic driving force is low, leading to less than 1% conversion that can be achieved. To achieve a significantly high conversion in this temperature range, the dehydrogenation reaction must be carried out in an atmosphere of inert gases such as nitrogen, reducing the partial pressure of H2 and shifting the equilibrium towards the product direction. In this case, the gaseous product is not pure H2, but H2 is highly diluted in inert gases. For practical industrial applications of H2, diluting H2 to concentrations that can be neglected is almost useless.
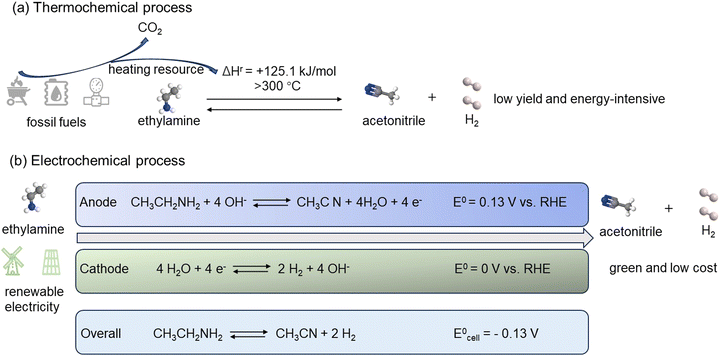 |
| | Fig. 2 (a) Traditional thermochemical process of ethylamine dehydrogenation. (b) Renewable electrochemical process of EDH coupling with HER. | |
On the contrary, utilizing environmentally friendly electric energy can achieve ethylamine dehydrogenation through an electrochemical process under mild conditions. The electric energy required for electrolyzing ethylamine into acetonitrile and H2 can be sourced from renewable energies such as solar or wind power, thereby reducing reliance on fossil fuels and mitigating environmental impacts. Furthermore, the thermodynamic potential required for the EDH half-cell reaction is only 0.13 V vs. RHE. Therefore, by coupling the electrochemical EDH with HER, ethylamine can be electrolyzed into acetonitrile and H2 with as low as 0.13 V cell voltage, much lower than the minimum 1.23 V required for water electrolysis (Fig. 2(b)). This not only can reduce energy consumption to promote efficient H2 production, but also facilitate the electrochemical synthesis of higher value-added acetonitrile.
2.3 Reaction mechanisms
In the 1980s, M. Trenary et al. investigated the reactivity and selectivity of ethylamine thermal decomposition on the W (100)-(5 × 1)-C surface.45 They conducted their research using temperature-programmed reaction spectroscopy and isotopic labelling methods and proposed an EDH surface intermediate, identified as CH3CNH, which then generated acetonitrile, the primary product of EDH, through the breaking of the N–H bond. However, they did not provide spectroscopic evidence to support their findings. Following closely, G. A. Somorjai et al. utilized high-resolution electron energy loss spectroscopy (HREELS) to investigate the thermal EDH mechanism on Ni(111) within the temperature range of 300–350 K.46 They proposed the same EDH surface intermediate, CH3CNH. The CH2 group of ethylamine was believed to interact with the Ni surface atoms, and then the adsorbed ethylamine molecules initially formed CH3CNH2 through the cleavage of the C–H bond, then proceeded to CH3CNH through stepwise cleavage of the N–H bond, and finally yielded CH3CN (Fig. 3(a)). In a subsequent work,47 they further validated this reaction mechanism through molecular modelling calculations of binding energies. It is worth noting that M. Trenary et al. studied EDH on the Pt(111) surface through infrared spectroscopy and density functional theory (DFT) calculations.48 They discovered a surface intermediate, CCHNH2, which is completely different in structure from the work reported by G. A. Somorjai et al. on Ni(111). This is likely due to Ni and Pt having completely different catalytic structures and intrinsic electronic properties. In subsequent work,49 they provided a more detailed mechanistic study through extensive experiments and spectroscopic data. They proposed that ethylamine molecules stably adsorb onto the Pt(111) surface at temperatures ranging from 85 K to 300 K. Partial dehydrogenation occurs at 330 K, leading to the formation of the first stable surface intermediate, CCHNH2, which desorbs as CH3CN between 340 and 360 K. Upon heating above 420 K, the un-adsorbed CCHNH2 further dehydrogenates, resulting in the cleavage of the remaining C–H bonds to form the second surface intermediate, CCNH2. Further heating above 520 K leads to the dissociation of the C–C bond and one N–H bond of CCNH2 to generate HCN (Fig. 3(b) and (c)). This sharply contrasts with the findings of G. A. Somorjai et al., who proposed that, after acetonitrile desorption, only C and N atoms remain on the Ni(111) surface at temperatures above 350 K.
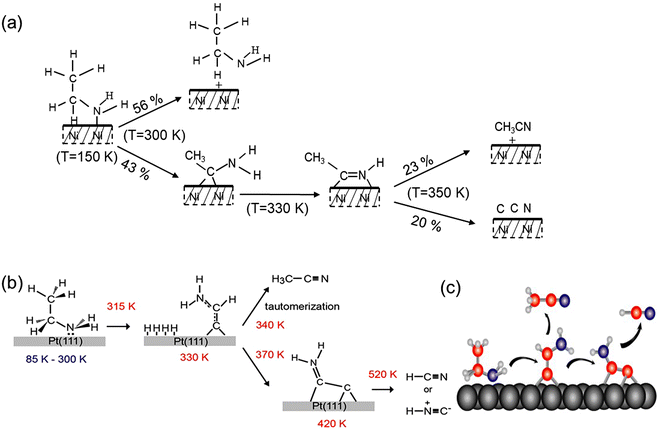 |
| | Fig. 3 (a) Thermal decomposition of ethylamine on Ni(111). Reproduced with permission from ref. 46. Copyright 1992 American Chemical Society. (b) Thermal decomposition process of ethylamine on Pt(111). (c) Schematic drawing of thermal decomposition of ethylamine on Pt(111). Reproduced with permission from ref. 49. Copyright 2013 American Chemical Society. | |
Unlike thermochemical processes, non-precious metal-based transition catalysts such as Ni-based and W-based catalysts have low thermodynamic redox potential. The surfaces of Ni-based and W-based catalysts would be already oxidized during electrochemical EDH, making it impossible to exist in stable metal-based catalytic forms. As early as 1999, F. Huerta et al.50 first studied the electro-oxidation behavior of ethylamine on Pt single-crystal electrodes in acidic media. They utilized cyclic voltammetry (CV) and Fourier-transform infrared reflection–absorption spectroscopy (FTIR) to investigate and found that ethylamine oxidation on the Pt electrode leads to the formation of adsorbed cyanide species, which act as poisons for further oxidation. On Pt(111), the adsorbed cyanide is quite stable; however, it is difficult to detect adsorbed cyanide on the Pt(110) surface; on Pt(100), the adsorbed cyanide exhibits high reactivity. The significant differences in the electro-oxidation of ethylamine on different crystal facets are likely due to variations in the adsorption capability of Pt electrodes for ethylamine, as well as substantial differences in the electronic structure and structural stability of different crystal facets. Unfortunately, they did not provide further evidence to prove the specific structural information of the adsorbed cyanide species. Similar to ammonia oxidation, there is no electrostatic repulsion between ethylamine molecules and electrolyte ions under alkaline conditions, leading to smaller mass transfer limitations. Therefore, EDH in alkaline media is more likely to occur than in acidic media. Our research group utilized in situ attenuated total reflection infrared spectroscopy (ATR-IR) and DFT calculations to investigate the EDH mechanism on Pt catalyst surfaces in alkaline media. As shown in Fig. 4(a), as the potential was increased, the rise of five peaks suggested that reaction species were formed and accumulated on the Pt surface. The peaks at 2969 cm−1 and 2875 cm−1 can be assigned to –CH3 stretching, and the other three peaks at 1635 cm−1, 1490 cm−1, and 1395 cm−1 can be assigned to –N–H bending mode.51 Notably, these peak positions were very similar to ethylamine adsorption, which suggested the structural similarity between the newly formed species and ethylamine. Based on these observations, we believe that the newly formed species on the Pt surface were EDH intermediates, i.e., partially dehydrogenated amines, such as CH3CHNH or other irregular structures, CH3CNH2, CH3CH2NH, CH3CNH, and CH3CHN. To better investigate the generation of reaction intermediates, the absorbance at 2969 cm−1 was chosen as an indicator of the concentration of reaction intermediates on the Pt surface. As shown in Fig. 4(b)–(d), the absorbance at 2969 cm−1 increased as the potential increased. This indicates the concentration of reaction intermediates increased with the potential, meaning the intermediates were being continuously generated and accumulated on the Pt surface as a function of potential and time. This is consistent with the oxidation behavior of Pt under acidic conditions reported by F. Huerta et al. To further understand the reaction pathway of EDH, we employed DFT modelling to calculate the free energy profile of the reaction (Fig. 4(e) and (f)). The results suggest that ethylamine molecules initially undergo spontaneous adsorption, forming the initial intermediate CH3CH2NH2*. The first dehydrogenation is a strongly uphill reaction, removing the H from the C–H bond adjacent to NH2 to generate CH3CHNH2*. Subsequently, the remaining H on the C–H bond adjacent to NH2 is removed to form CH3CNH2*. Then, the third dehydrogenation occurs on the N–H bond to produce CH3CNH*, followed by the loss of the final H on the N–H bond to generate CH3CN. Throughout the entire EDH, the fourth removal of the H from the N–H bond presents the highest energy barrier on Pt, indicating that breaking the last N–H bond is the rate-determining step of the entire reaction. It is worth mentioning that this result is quite different from the thermal catalytic mechanism of ethylamine on Pt(111) reported by M. Trenary et al. It is well understood that thermal catalysis and electrocatalysis exhibit significant differences in catalytic mechanisms due to variations in energy sources, reaction kinetics, and reaction conditions. At present, there is still limited research on the electrochemical EDH, and the specific mechanism remains inconclusive. More approaches are needed to carefully identify the generation of intermediates, which is crucial for mechanistic catalysis understanding and for the design of high-performance EDH electrocatalysts.
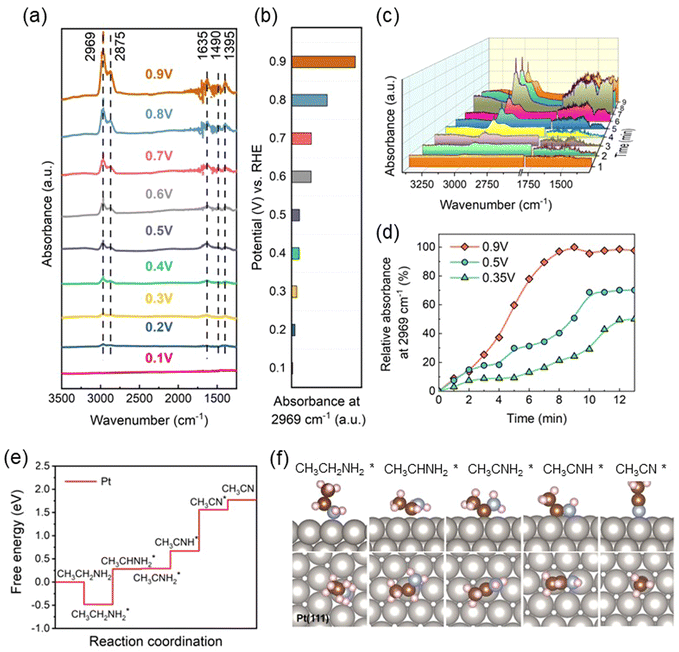 |
| | Fig. 4 (a) ATR-IR spectra on Pt in 1 M CH3CH2NH2 + 1 M KOH solution at various potentials from 0.1 V to 0.9 V vs. RHE. (b) The absorbance on Pt surface at 2969 cm−1 and various potentials from 0.1 V to 0.9 V vs. RHE. (c) Time resolved ATR-IR spectra of Pt surface at 0.9 V vs. RHE. (d) Relative absorbance of pure Pt at 2969 cm−1 at 0.35 V, 0.5 V, and 0.9 V vs. RHE with respect to time. (e) Calculated free energy diagram for EDH. (f) Adsorption configurations of intermediates at Pt surface. | |
3. Electrocatalysts in ethylamine dehydrogenation
According to the current research status, EDH electrocatalysts can be divided into two main categories: PGM and non-PGM catalysts. This section briefly reviews recent research achievements from the perspective of catalytic active sites, aiming to provide insights into the design of novel EDH electrocatalysts.
3.1 PGM catalysts
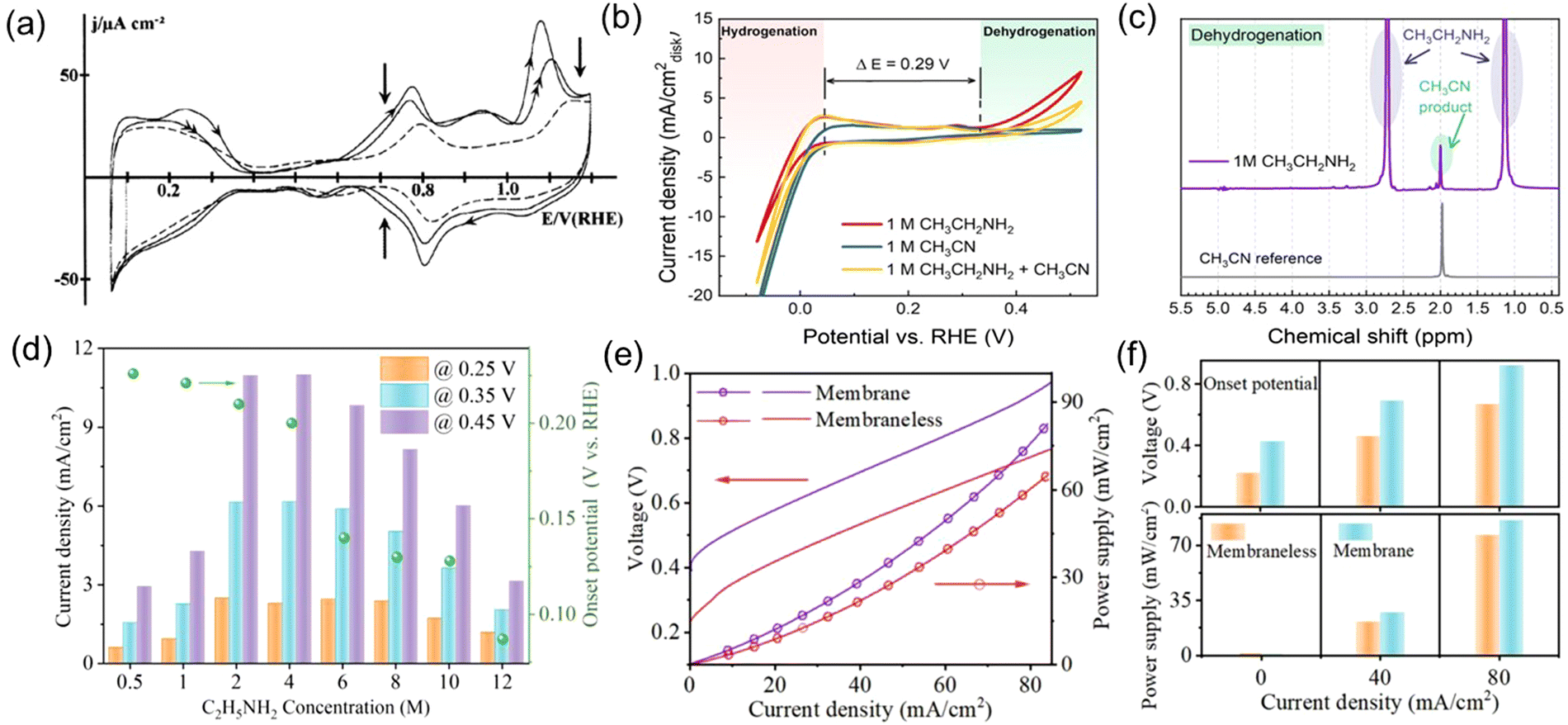
At present, research has focused on Pt-based catalysts, owing to their EDH reactivities. As discussed before, F. Huerta et al.50 studied the electro-oxidation behavior of ethylamine on Pt(111) electrodes in acidic media. The CV curve showed poor catalytic activity for EDH, with 0.6 V vs. RHE high onset potential and a low peak current density of 50 μA cm−2 (Fig. 5(a)). This indicates the slow reaction kinetics of EDH on Pt electrodes in acidic media, which is probably due to the positively charged ethylammonium formation in acidic media, leading to repulsion with electrode surface and thus hindrance in mass transfer. In this case, our research group investigated the electrochemical EDH performance of Pt catalyst under alkaline conditions.22 The fast kinetics of EDH were demonstrated by an anodic onset potential of only 0.32 V vs. RHE and a peak current density of 10 mA cm−2 (Fig. 5(b)). Moreover, acetonitrile was identified as the sole liquid-phase product, indicating the high selectivity of Pt catalysts for EDH to acetonitrile (Fig. 5(c)). In another study, we investigated the influence of ethylamine concentration on its dehydrogenation performance, as well as the performance of anion exchange membrane (AEM) electrolyzers and membraneless electrolyzers coupling EDH with HER.21 The EDH rate initially increased and then decreased with increasing ethylamine concentration, reaching maximum current density at 2 M ethylamine concentration (Fig. 5(d)). This indicates that relatively high ethylamine concentrations promote the EDH rate. Still, excessively high concentrations become disadvantageous, probably due to the limited solubility of ethylamine in alkaline media, leading to hindered mass transfer and, ultimately, slower electrochemical processes. The electrolysis performance of ethylamine in AEM electrolyzers and membraneless electrolyzers indicates that membraneless cells have lower internal resistance, resulting in better cell performance and higher energy efficiency (Fig. 5(e) and (f)). The poorer performance of AEM electrolyzers may be attributed to the plasticizing effect of the polymer main chain of the AEM.
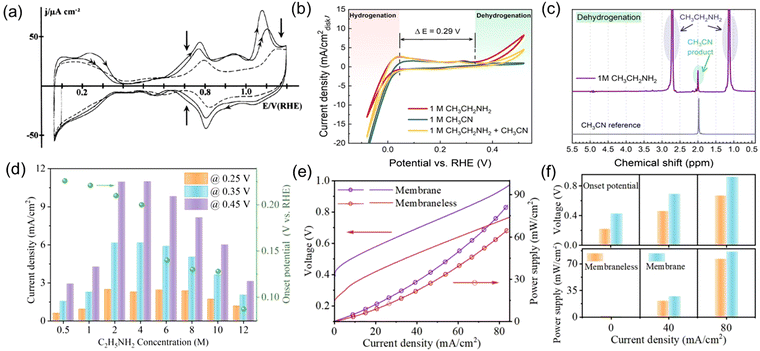 |
| | Fig. 5 (a) CV curve for a Pt(111) electrode immersed in 0.1 M HClO4 + 0.1 M CH3CH2NH2 solution. Reproduced with permission from ref. 50. Copyright 1999, Elsevier. (b) CV of commercial Pt black catalyst-loaded working electrode in 1 M NaOH with the addition of 1 M CH3CH2NH2, 1 M CH3CN, and 1 M CH3CH2NH2 + 1 M CH3CN. (c) 1H NMR spectrum of the liquid product from CH3CH2NH2 dehydrogenation obtained by applying 0.7 V vs. RHE for 10 h. Reproduced with permission from ref. 22. Copyright 2021 American Chemical Society. (d) The influences of ethylamine concentration on the EDH onset potential and current density. (e) Polarization curves and power supply profiles using AEM and membraneless ethylamine electrolysis cells. (f) Performance comparison of the two cells regarding cell onset voltage and needed power supply at different current density. Reproduced with permission from ref. 21. Copyright 2023, Elsevier. | |
Even though the high catalytic activity of Pt catalysts for EDH has been demonstrated and reported, these catalysts may initially be effective, but they often experience gradual deactivation during long-term operation. The deactivation of the catalyst occurs through the adsorption of nitrile-like intermediates onto the Pt surface, forming strong interactions that hinder desorption, thereby blocking catalytic active sites and causing gradual deactivation. (Fig. 6(a)). Interestingly, in our research we found that when the CV scan range was enlarged negatively to −0.3 V vs. RHE, the current retention was nearly 100% after 10 consecutive scans, considerably higher than that of the 0–0.9 V vs. RHE scan, which suggests that the generated reaction intermediates on Pt surface could be completely removed, exhibiting a self-cleaning process, leads to a remarkable stability of the Pt catalyst without significant activity decay (Fig. 6(b)). In situ ATR-IR spectrum is a valuable tool for providing evidence of this self-cleaning process. Fig. 6(c) shows the evolution of the ATR-IR spectrum at selected positions during a complete CV cycle. Reaction intermediates started to form when the CV is swept positively, and the concentration on the Pt surface reached its maximum signal at 0.9 V vs. RHE, evidenced by the highest absorbance at 2969 cm−1. With the change of scan direction, the concentration of the intermediates decreased slightly towards lower potentials. When the potential was swept in the hydrogen underpotential deposition (H-upd) region and HER region, protons were deposited on the Pt surfaces, which would facilitate the intermediates being hydrogenated back to ethylamine. Therefore, in the lower potential range, the intermediate hydrogenation and the removal from the Pt surface were greatly promoted, which can be confirmed by the absorbance at 2969 cm−1 disappearing at −0.2 V vs. RHE. However, the most effective approach to enhance the long-term stability of Pt catalysts remains a deep understanding of the catalytic mechanism and designing from the ground up. For instance, one effective strategy is to rational design binary or ternary alloys. Such synergistic interactions can modify the electronic structure of Pt as well as the surface geometry, weakening its ability to adsorb intermediates, thus facilitating the removal of intermediates and the regeneration of Pt metal catalytic active sites. Therefore, our group synthesized Pt–Ni alloy nanoparticles with various compositions to improve the activity and stability for EDH. Pt5Ni/C, Pt3Ni/C, and PtNi/C with metal loading of 20 wt% were synthesized using a modified solid chemistry method. Fig. 6(d) shows the CV curves of Pt5Ni/C, Pt3Ni/C, PtNi/C and Pt/C electrocatalysts in 1 M CH3CH2NH2 + 1 M KOH solution. The current density was normalized to the surface area of Pt to evaluate the specific activity of the catalyst. Among these catalysts, the Pt3Ni/C alloy nanoparticles showed the highest EDH activity and improved stability (Fig. 6(e)). The reasons for the enhanced activity and stability of Pt–Ni alloy can be attributed to that Pt3Ni has a lower d-band center compared to pure Pt, which causes the intermediates to bind more weakly to the Pt active sites. Besides, the adsorption of the reaction intermediates may require multiple anchoring points. Incorporating a second element into the Pt lattice can disrupt Pt ensembles, diminishing the likelihood of intermediates locating ample anchoring points.
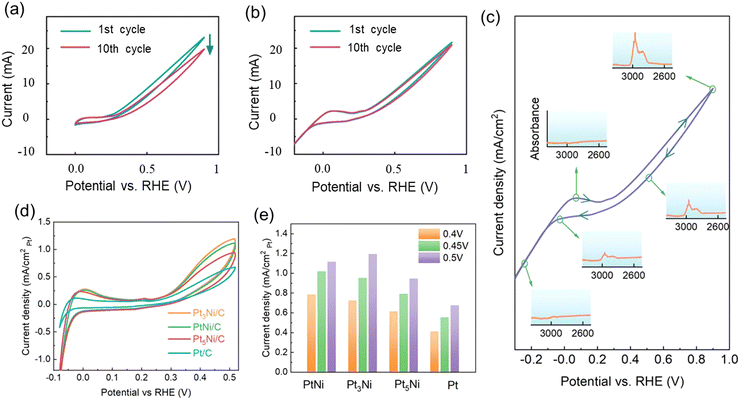 |
| | Fig. 6 (a) First and tenth CV curves of pure Pt in the range of 0 V to 0.9 V vs. RHE. (b) First and tenth CV curves of pure Pt in the range of −0.3 V to 0.9 V vs. RHE. (c) In situ ATR-IR spectroscopy and absorbance evolution within a CV scan of pure Pt. (d) CV curves of PtNi/C, Pt3Ni/C, Pt5Ni/C, Pt/C catalysts in 1 M CH3CH2NH2 + 1 M KOH solution. (e) Current density of EDH on PtNi/C, Pt3Ni/C, Pt5Ni/C, Pt/C catalysts at 0.4 V, 0.45 V, and 0.5 V vs. RHE. | |
In addition, the ethylamine electro-oxidation performance on Au electrodes have also been investigated. In 1995, D. C. Johnson et al.52 studied the electrochemical behavior of ethylamine on Au electrodes in alkaline media through a combination of linear sweep voltammetry (LSV) and pulsed electrochemical detection (PED) on a RRDE electrode. The oxidation of ethylamine occurs concurrently with the formation of surface oxide species (Au → AuOH + AuO). However, the AuO is inactive for further ethylamine oxidation. They proposed that the adsorption of ethylamine occurs through two mechanisms, i.e., irreversible chemisorption and reversible co-adsorption with OH−. Moreover, they suggested that the probable product of ethylamine oxidation, acetonitrile, is strongly adsorbed to the Au electrode, leading to surface poisoning of the Au electrode. This necessitates potential excursions into regions for oxide formation and reduction to induce the desorption of acetonitrile and reactivate the electrode surface. This is consistent with the poisoning mechanism observed on Pt electrodes.
3.2 Non-PGM catalysts
In non-PGM-based catalysts, research mainly focuses on transition metal oxides and hydroxides, such as NiO/Ni(OH)2. However, compared to Pt-based catalysts, the EDH performance of non-PGM catalysts is evidently inadequate. The primary reason is their lower affinity for ethylamine, resulting in higher EDH overpotentials and slower reaction kinetics. Weidong Ao and colleagues investigated the ethylamine oxidation performance over Pd–Ni(OH)2 heterostructure nanosheet.53 As shown in Fig. 7(a), the onset potential for ethylamine electro-oxidation on Pd–Ni(OH)2 nanosheets is approximately 1.32 V vs. RHE, significantly higher than that on Pt-based catalysts. Additionally, the anodic current density obtained on Pd–Ni(OH)2 nanosheets at the same potential is higher than that on Pd nanosheets and Ni(OH)2 nanosheets. This indicates that Ni-based compounds serve as primary active sites for the ethylamine oxidation reaction, which is why we classify Pd–Ni(OH)2 as a non-PGM catalyst. However, Pd exhibits poor performance in ethylamine oxidation, likely due to the difficulty of desorbing the generated acetonitrile, which occupies the active sites of the Pd catalyst, which could be a common problem of PGM catalysts. Fig. 7(b) shows excellent long-term electrocatalytic stability of Pd–Ni(OH)2 at different voltages, completely different from Pt-based catalysts. This is because EDH on Pt-based catalysts and Ni-based catalysts follows different catalytic mechanisms. On Pd–Ni(OH)2, the EDH follows a reducible oxide catalysis mechanism, where NiIII/NiIV species are in situ formed on the electrode surface, serving as actual active sites for EDH. The high valence of the metal center enhances electron acceptance and facilitates faster charge transfer rates, effectively promoting the direct oxidation of adsorbed ethylamine to acetonitrile. Like Pt-catalyzed EDH, acetonitrile is the sole liquid product of ethylamine catalyzed by Pd–Ni(OH)2 (Fig. 7(c)). They also utilized DFT calculations to investigate the dehydrogenation mechanism. Ethylamine first adsorbs onto the surface of the Pd–Ni(OH)2, then gradually oxidizes to *CH3CH2NH, *CH3CHNH, *CH3CHN, *CH3CN, and finally desorbs as CH3CN (Fig. 7(d)–(f)). The breaking of the N–H bond requires a higher energy barrier, especially the breaking of the first N–H bond, which is the rate-determining step of the entire reaction. The construction of the Pd–Ni(OH)2 heterojunction lowers the reaction energy barrier, thereby improving the reaction kinetics and catalytic performance.
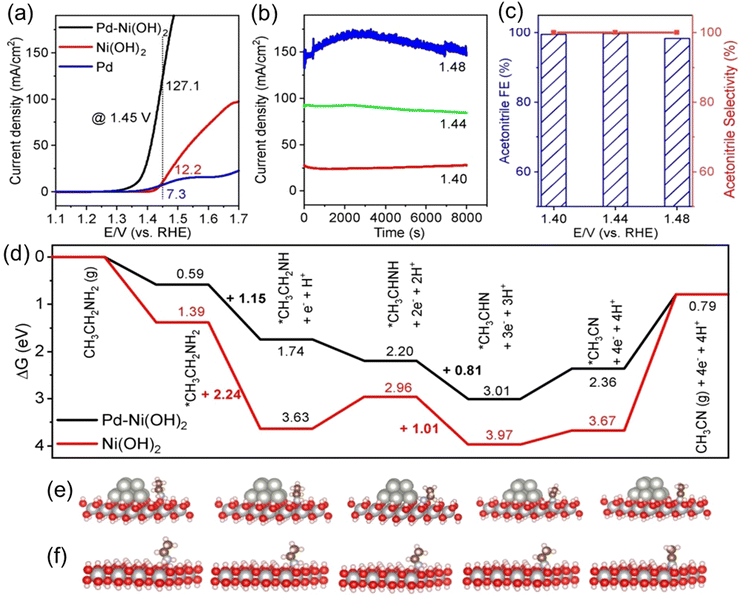 |
| | Fig. 7 (a) Electrode surface normalized polarization curves in 1.0 M KOH + 0.5 M CH3CH2NH2 aqueous solution on Pd–Ni(OH)2 NSs, Pd NPs and Ni(OH)2 NSs. (b) Chronoamperometric current at different potentials for 8000 s. (c) Faradaic efficiencies of CH3CN at different potentials. (d) Free energy diagrams for CH3CH2NH2 electro-oxidation to CH3CN on Ni(OH)2 and Pd–Ni(OH)2 at an applied potential U = 0 V. (e) The adsorption models of different oxidized intermediates on Pd–Ni(OH)2. (f) The adsorption models of different oxidized intermediates on Ni(OH)2. Reproduced with permission from ref. 53. Copyright 2023, Wiley-VCH. | |
4. Applications of electrocatalytic ethylamine dehydrogenation
The transportation and energy conversion, based on energy carriers, are crucial for meeting global energy demand and achieving energy sustainability. Ethylamine, as a high hydrogen content energy carrier, can play a significant role in the field of energy storage and conversion utilizing EDH. In this section, we will discuss the potential applications of EDH in H2 storage, regenerative fuel cells, and batteries. While there have been reports on H2 storage, so far, the application in regenerative fuel cells and batteries has not been reported. Therefore, to promote research in this field, we will also discuss its potential applications in regenerative fuel cells and batteries, in addition to hydrogen storage.
4.1 Hydrogen storage
In recent years, the use of LOHC for H2 storage has garnered increasingly widespread attention due to its advantages of achieving sustainable, convenient, and efficient H2 storage and transportation with no CO2 emissions and other environmental pollutants. Due to the endothermic nature of the dehydrogenation reaction requiring higher temperatures and pressures, electrochemical LOHC systems possess more pronounced advantages compared to thermochemical LOHC systems. Moreover, electrochemical LOHCs offer more efficient, safer, and lower temperature and pressure for H2 storage and utilization solutions. Compared to alcohol-based LOHCs like isopropanol (IPA), the reversible EDH to acetonitrile for H2 storage is particularly attractive because each ethylamine releases two H2 molecules. In our previous work,22 we reported a novel, electrochemical ethylamine/acetonitrile redox method for efficient H2 storage with a theoretical storage capacity of 8.9 wt% at completely ambient conditions, surpassing the 2025 DOE onboard H2 storage target. The H2 uptake process was realized by H2 oxidation reaction (HOR) on the anode and AHD on the cathode, where acetonitrile is electrochemically hydrogenated to ethylamine. The H2 release process was realized by HER on the cathode and EDH on the anode, where ethylamine is electrochemically dehydrogenated to acetonitrile. Fig. 8(a) illustrates a sustainable H2 economy blueprint via the ethylamine/acetonitrile redox pairs for H2 storage and transportation. H2 can be generated utilizing renewable energy sources such as solar or wind power through water electrolysis. The produced H2 is stored via AHD, with the generated ethylamine serving as a hydrogen carrier that can be transported directly through existing petroleum and natural gas pipelines. Finally, ethylamine is electrochemically dehydrogenated at the terminal for on-demand uses, for instance, at hydrogen stations or fuel cells. Chronoamperometry tests showed consistent current generation, even at low cell voltages, indicating good durability for AHD (Fig. 8(b)). During EDH process, the generation rate of H2 increased rapidly with voltage, exhibited a high faradaic efficiency (FE) close to 100% (Fig. 8(c)). The high FE of EDH and the sustainability of AHD demonstrate the potential of ethylamine/acetonitrile redox pairs for efficient H2 storage. Besides, this method demonstrates significantly lower energy consumption compared to energy generation via H2 combustion (Fig. 8(d)). Fig. 8(e) shows the summarized data of the comparison between the electrochemical ethylamine/acetonitrile redox method and other traditional methods reported in the literature for H2 storage. It is evident that the electrochemical ethylamine/acetonitrile redox method stands out for its ability to achieve a high storage capacity under ambient conditions, which makes it a feasible H2 storage strategy, capable of fostering the advancement of the H2 economy and conducive to propelling the development of H2 economy to a new level.
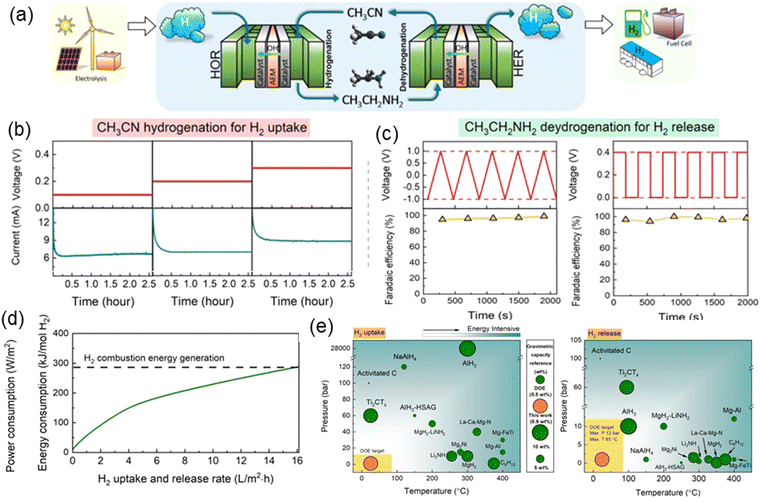 |
| | Fig. 8 (a) A blue print based on ethylamine/acetonitrile that realizes a complete cycle of H2 production, storage and application under ambient conditions. (b) Hydrogenation of acetonitrile for H2 uptake under various cell voltages and the measured ethylamine generation rate. (c) Chronoamperometry test of ethylamine dehydrogenation for H2 release, and the measured H2 generation FE. (d) Calculated energy consumption for completing a complete H2 storage cycle as a function of H2 uptake and release rate. (e) Comparison of the electrochemical ethylamine/acetonitrile redox method with other reported methods in terms of H2 storage capacity, operation pressure, and temperature. Reproduced with permission from ref. 22. Copyright 2021 American Chemical Society. | |
4.2 Regenerative fuel cells
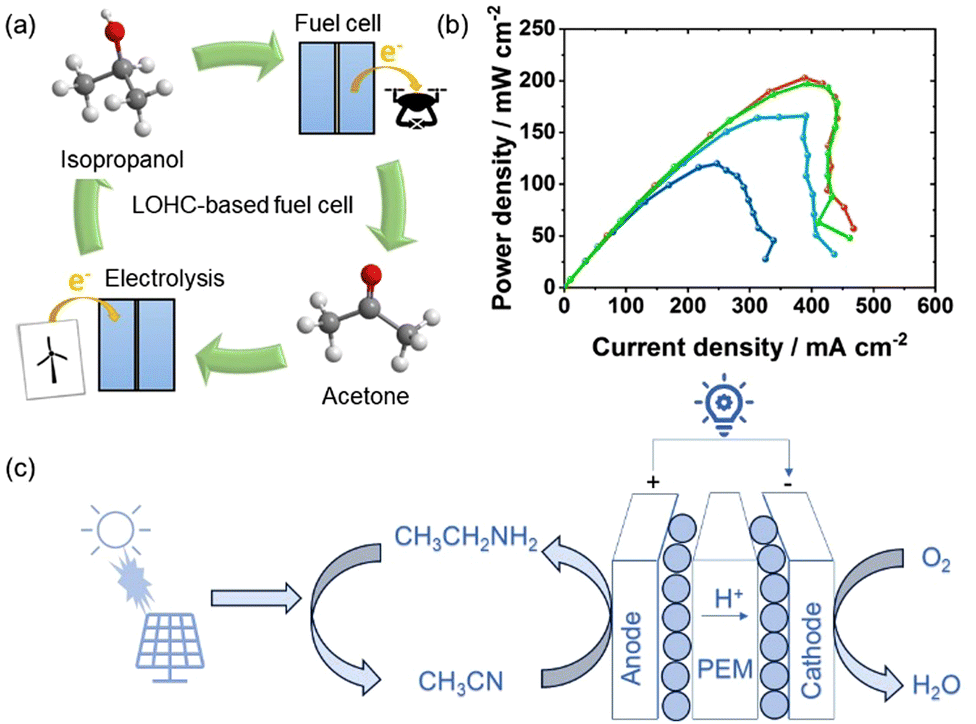
The intensifying energy shortage crises have catalyzed sustainable development and hastened the adoption of fuel cell technology, which generates environmentally friendly electrical energy through highly efficient and low-emission electrochemical reactions. Over recent decades, the practical integration of fuel cell vehicles into the automotive market has propelled extensive research in this domain. Direct liquid fuel cells (DLFCs), engineered to address fuel storage and safety concerns, have emerged as a pivotal focus within fuel cell research.54,55 Various carbon-based DLFCs, including the direct methanol fuel cell (DMFC), direct formic acid fuel cell (DFAFC), and direct ethanol fuel cell (DEFC), have undergone rigorous investigation. In particular, the DMFC has garnered significant attention owing to its exceptional fuel energy density and stability, facilitating convenient and efficient long-distance transportation and storage, and the capability to achieve high-power densities for effective on-demand electricity generation.56,57 However, these alcohol-based fuel cells are non-renewable and contribute to CO2 emissions.58 In this context, the concept of LOHC-based regenerative fuel cells has emerged.59 Hydrogen-rich LOHC compounds can be used directly as fuel, with LOHC dehydrogenation and fuel cells operating in the same device, requiring only one catalyst, which is advantageous for saving space and reducing costs. In addition, H2 is transferred to organic carrier liquids that can directly undergo hydrogenation, allowing for the regeneration of hydrogen-rich LOHC fuel in this way, with no CO2 emissions throughout the entire process. In 2020, Pascal et al. reported IPA-based LOHC regenerative fuel cells.60Fig. 9(a) illustrates the schematic of the LOHC system utilizing IPA and acetone for H2 storage and fuel cells, with the direct utilization of LOHC-bound H2 in a low-temperature PEM fuel cell being particularly attractive. This fuel cell demonstrates a power density of 203 mW cm−2 at an absolute pressure of 300 kPa, which is 20% of that achieved by a pure hydrogen fuel cell and twice that of the most advanced DMFC (Fig. 9(b)). Although using IPA/acetone as an energy storage system is visionary, incomplete oxidation of IPA in fuel cells is disadvantageous, as each fuel molecule only exchanges two electrons. Moreover, IPA has certain limitations, such as a H2 storage capacity of only 3.3 wt% H2 or 25.9 gH2 L−1, which are significantly lower than ethylamine in terms of H2 storage capacity of 8.9 wt% H2 or 72.1 gH2 L−1. Besides, the thermodynamic theoretical open circuit voltage of IPA fuel cells under standard conditions is 1.07 V,61 slightly lower than the theoretical open circuit voltage of ethylamine fuel cells of 1.1 V. Therefore, compared to IPA-based fuel cells reported to date, ethylamine fuel cells exhibit unique advantages. Fig. 9(c) depicts the fundamental principle behind ethylamine-based regenerative fuel cells, showcasing that EDH occurs on the anode and oxygen reduction reaction (ORR) occurs on the cathode. These electrochemical processes serve as the cornerstone of regenerative fuel cells, facilitating electricity generation while transforming ethylamine into acetonitrile. Acetonitrile, in turn, readily reacts with renewable hydrogen resources, regenerating ethylamine for subsequent use. The impressive process makes ethylamine a highly efficient regenerative fuel and distinguishes it from almost all other direct liquid fuels that have achieved reasonable power generation in fuel cells. Furthermore, compared to many existing non-CO2 generating alternative fuels, like cyclohexane/benzene, the ethylamine/acetonitrile LOHC system can not only produces more power in fuel cells but exhibits low chemical toxicity. Although it is evident that ethylamine-based LOHC regenerative fuel cell is advantageous, there is still much space for further improvement. For example, in order to enhance fuel cell performance, the development of superior anode catalysts and the engineering of membrane electrode assemblies (MEAs) for optimum operation are required.
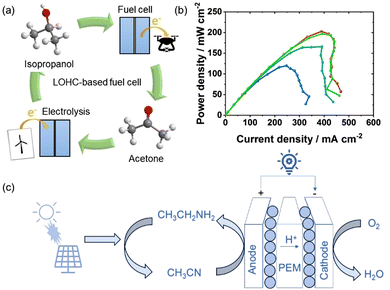 |
| | Fig. 9 (a) Schematic of IPA-based LOHC fuel cell system. (b) Power density curves of the IPA-based LOHC fuel cell under 85 °C and varying pressure conditions (100–400 kPa absolute). Reproduced with permission from ref. 60. Copyright 2020, Elsevier. (c) Diagram of ethylamine-based LOHC regenerative fuel cells strategy for emission-free electrical energy storage and generating system. | |
4.3 Batteries
The storage and release of energy are the main obstacles to the widespread use of intermittent renewable energy sources such as wind and solar energy. Electrochemical energy storage devices such as batteries offer hope for achieving sustainable energy supply. In recent years, aqueous batteries have stood out among various energy storage devices due to their high safety and the conducive high ionic conductivity of aqueous solutions for rapid redox reactions in rechargeable batteries.62 In particular, aqueous rechargeable hydrogen gas batteries have been actively researched due to their advantages, such as low overpotential, fast kinetics, high energy density, and strong stability of the hydrogen gas anode.63 However, the application of hydrogen gas batteries for energy storage is hindered by challenges in H2 storage and transportation. Therefore, to promote hydrogen battery technology, the development of LOHC batteries would be ideal to overcome those disadvantages. Shibo and colleagues reported a hydronium-ion battery utilizing a soluble methylene blue (MB) anode and a MnO2@graphite felt cathode (Fig. 10(a)).64 The charge and discharge of this battery rely on the –CN/–C–N–H group transition at the anode and MnO2/Mn2+ transition at the cathode. As seen in Fig. 10(b), from the perspective of practical applications, the charge and discharge capacity achieved by this battery is too low, so there is still a long way to go before it can be applied effectively. In our recent research,65,66 we presented the concept of a LOHC battery utilizing cyclohexanol (CHOL) and IPA as anode materials. These materials exhibit excellent reversible and selective interconversion with their redox counterparts under Pt catalysis. Coin cells were assembled under ambient conditions using commercial Pt/C and Ni(OH)2/NF as the anode and cathode materials, respectively (Fig. 10(c)). We demonstrated that the CHOL-based cell exhibited a charge capacity of 374 mA h g−1 at 1C with 90% efficiency over 300 hours, and the IPA-based cell exhibited a charge capacity of 525 mA h g−1 at 1C with 95% efficiency. In Fig. 10(d), we compared the voltage and specific capacity of hydrogen gas batteries or proton batteries reported in the literature with CHOL and IPA-based LOHC batteries. CHOL and IPA-based LOHC batteries demonstrate significantly higher specific capacities and cell voltages, proving the advantages of LOHC batteries. However, compared to ethylamine, CHOL and IPA exhibit substantially lower mass and volumetric hydrogen carrier densities. Therefore, benefiting from ethylamine's high hydrogen carrier content, replacing IPA with ethylamine as the anode material in LOHC batteries will offer greater advantages in terms of charge–discharge capacity. Fig. 10(e) illustrates a rough schematic of an ethylamine-based LOHC battery, if we disregard practical operating conditions, losses, and other limiting factors, the theoretical specific capacity of the ethylamine is 2382 mA h g−1, which means that in ideal conditions, each gram of ethylamine can store 2382 mA h−1 of charge. In Fig. 10(f), we compared the theoretical energy capacity that can be achieved by the present battery and other organic compound. The energy capacity that ethylamine can obtain is much higher than that of existing batteries and other compounds. This numerical value is more than 10 times higher than the theoretical energy density of the currently dominant lithium-ion batteries, being cheaper and safer as well, making it a promising power source. However, to some extent, the development of EDH electrocatalysts is at the core of the electrochemical conversion process. The properties of electrocatalysts determines the overall performance and efficiency of related devices to a certain extent. Therefore, the development of cost-effective EDH electrocatalysts with high activity and stability will still be the key point for the widespread application of this LOHC energy conversion technology.
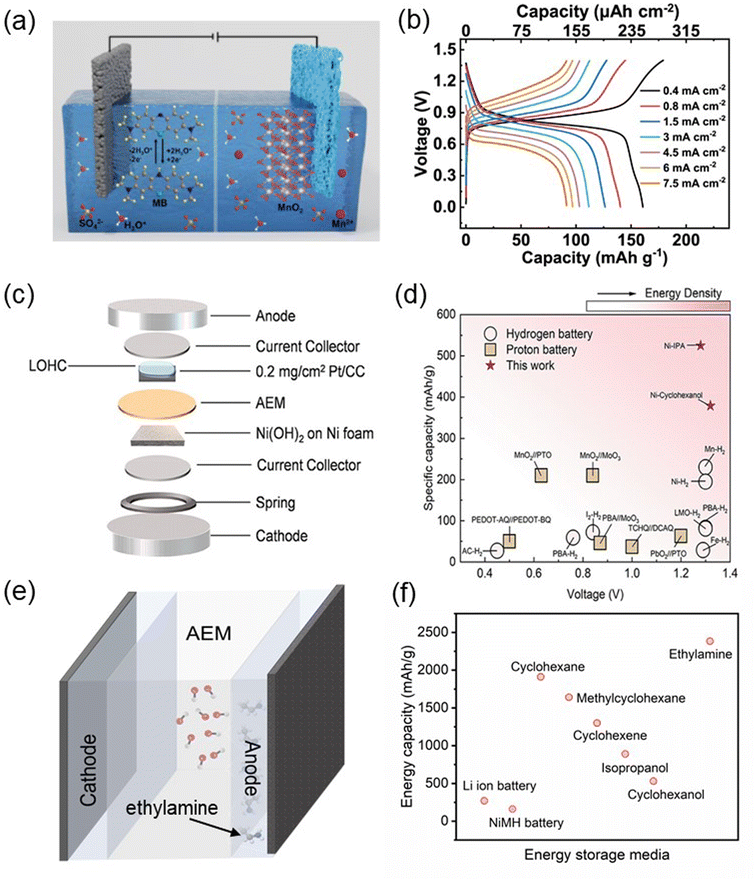 |
| | Fig. 10 (a) Schematic illustration of operation mechanism of the MB//MnO2 hydronium-ion batteries. (b) Charge and discharge curves of MB//MnO2 hydronium-ion battery at various current density. Reproduced with permission from ref. 64. Copyright 2024, Elsevier. (c) Schematic drawing of the assembly of the coin cell based on Ni(OH)2/NF-LOHC/Pt/CC batteries. (d) Comparison of the voltage and specific capacity of Ni(OH)2/NF-LOHC/Pt/CC batteries to other hydrogen batteries and proton batteries. Reproduced with permission from ref. 65. Copyright 2023 American Chemical Society. (e) A rough schematic illustration of the ethylamine-based LOHC batteries. (f) Energy capacity for power supply of present batteries and various organic compounds. | |
5. Conclusions and outlook
In summary, the electrocatalytic EDH is an energetically economical half-cell reaction that can play a significant role in the field of energy storage and conversion. In this feature article, we systematically summarized the fundamentals of thermocatalytic and electrocatalytic EDH. We specifically highlighted the recent research progress and critical issues regarding electrocatalytic EDH, particularly focusing on two types of electrocatalysts (Pt-based metals and Ni-based hydroxides), many of which are our research findings. Additionally, we discussed the application prospects of electrocatalytic EDH in energy storage and conversion, especially in H2 storage, regenerative fuel cells, and batteries. However, despite some inspiring progress, several urgent issues still need to be further researched.
5.1 Mechanistic study
Designing better electrocatalysts for EDH relies heavily on a detailed understanding of the mechanism. As a four-electron reaction, EDH involves multiple intermediates, leading to a complex reaction mechanism. However, there is currently limited experimental research and theoretical calculations on electrocatalytic EDH, and the reaction mechanism still remains inconclusive. Therefore, it is essential to further elucidate its catalytic mechanism. Nevertheless, due to the lack of effective experimental means and direct evidence to determine the unstable intermediates in the catalytic process, the study of reaction mechanisms has always been a challenge in the field of catalysis. In this case, by utilizing the existing experimental and theoretical analysis methods to elucidate plausible catalytic mechanisms, we can better understand the reaction pathway and provide a theoretical basis for catalyst design. Currently, the main method for studying EDH in electrochemistry is cyclic voltammetry, but this method is insufficient to fully elucidate the entire catalytic process. Therefore, supplementing more experimental and theoretical studies, especially on the investigation of reaction intermediates, is essential for a deeper exploration of catalytic mechanisms. Firstly, we suggest utilizing operando spectroscopy to better understand catalyst functioning and catalytic mechanisms. For instance, employing in situ FTIR spectroscopy to monitor real-time chemical bond changes and intermediate formation during catalytic reactions under different electrode potentials and reaction times, investigating the interactions between catalysts and reactants, as well as surface modification and reconstruction phenomena occurring during the reaction process. Using in situ Raman spectroscopy to monitor real-time structural changes on the catalyst surface during catalytic reactions can help understand the active sites of catalysts, reaction mechanisms, and interactions between catalysts and reactants, providing direct and dynamic evidence for the structure–property relationship of catalysts and guiding the design of more efficient catalysts. Secondly, DFT calculations play a crucial role in studying catalytic mechanisms. DFT can complement experimental studies by providing atomic-level understanding and predictive capabilities. For example, calculating reaction energies, activation energies, and reaction pathways helps understand the thermodynamic and kinetic properties of catalytic reactions. By simulating the interactions between reactants, intermediates, and catalyst surfaces, DFT can elucidate possible detailed pathway of catalytic reactions, including bond-breaking and bond-forming steps. Additionally, machine learning may become an auxiliary tool for mechanism study. Through machine learning algorithms, processing and analyzing large amounts of experimental data and computational results can help identify critical factors and essential parameters in catalytic reactions, assist in determining complex catalytic reaction mechanisms, and provide new insights and understanding.
5.2 Catalyst design
As discussed in the previous section, the main electrocatalysts for EDH are focusing on Ni-based hydroxides and Pt-based metal catalysts to date. On the one hand, Pt-based catalysts exhibit excellent activity for EDH, but even if we exclude their expensive cost, they still suffer from the fatal limitation of rapid deactivation due to poisoning during the catalytic process. On the other hand, researchers have also investigated some non-precious metal catalysts to replace the expensive and rapidly deactivating Pt-based catalysts, primarily focusing on Ni-based oxide or hydroxide electrocatalysts. However, their activity still significantly lags behind that of Pt-based catalysts. Additionally, they typically require the employment of high electrode potentials, and these catalysts form in situ high-valence catalytic active species at high potentials to ensure effective EDH and good stability. Nevertheless, the high overpotential of Ni-based electrocatalysts will unavoidably lead to unnecessary side reactions and lower energy efficiency. For the next generation of EDH systems, it is essential to design highly efficient electrocatalysts with a well-balanced activity (low onset potential) and stability (low catalyst deactivation rate). Considering that the two main methods to enhance catalyst activity are the geometric and electronic effects, we suggest focusing on catalyst design from these two aspects. For Pt-based catalysts, the focus lies on improving their stability during long-term operation. According to current research conclusions, the rapid deactivation of Pt-based catalysts is attributed to the strong adsorption of intermediate species on the catalyst surface. Therefore, based on the d-band center theory, on one hand, we suggest investigating the alloying effect. This involves designing Pt-based binary, ternary, or even multinary high-entropy alloys. By controlling the crystal phase and electronic structure through interactions between different elements, optimizing electron transfer pathways and the adsorption strength of reaction intermediates on the catalyst surface, thereby catalyst activity and stability can be enhanced. Simultaneously, for the Pt-based catalyst alloying strategy, we suggest conducting precise experimental control and detailed characterization of chemical compositions, such as utilizing advanced materials characterization techniques like X-ray absorption fine structure (XAFS), X-ray absorption near edge structure (XANES), and X-ray photoelectron spectroscopy (XPS). Furthermore, reliable experimental results can serve as a basis for structural simulation and related theoretical calculations to determine the influence of variable chemical compositions on the local electronic structure. It is well known that the structure of materials often significantly influences their performance. However, structure and facet control strategies have not yet been reported for EDH on Pt-based catalysts. Therefore, on the other hand, we suggest starting with the design of new Pt-based catalysts from aspects such as support, size, structure, and facet control. Ni-based hydroxide catalysts follow different reaction mechanisms and pathways compared to Pt-based catalysts. The key to enhancing their catalytic activity and efficiency lies in reducing their onset oxidation potential, but research in this area remains very limited. We still recommend starting from electronic structure and geometric structure control, such as morphology and size control, oxide support or carbon support control, as well as strategies involving composite with other materials or doping.
Ultimately, these bottlenecks indicate that there is still significant research space for the half reaction of EDH, we believe that this feature article can inspire research in the fundamentals of EDH, the design and optimization of electrocatalysts, and their applications in relevant energy fields.
Author contributions
Zhenmeng Peng conceived, organized, reviewed and edited the manuscript. Yanlin Zhu did literature research and wrote the original draft. Dezhen Wu provided part of the research data. Jinyao Tang provided help with manuscript editing. Dakota Braaten and Bin Liu performed part of the DFT calculations. All authors contributed to the completion of the manuscript.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge support from the University of South Carolina for this study.
Notes and references
- J. R. Reimers and L. E. Hall, J. Am. Chem. Soc., 1999, 121, 3730–3744 Search PubMed.
- Z. Liu, F. Huang, M. Peng, Y. Chen, X. Cai, L. Wang, Z. Hu, X. Wen, N. Wang, D. Xiao, H. Jiang, H. Sun, H. Liu and D. Ma, Nat. Commun., 2021, 12, 6194 Search PubMed.
- M. North, Angew. Chem., Int. Ed., 2005, 44, 2053–2055 Search PubMed.
- W. B. Vishwas, G. Chandrashekhar, M. Beller and R. V. Jagadeesh, Science, 2022, 376, 1433–1441 Search PubMed.
- D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2015, 357, 883–900 Search PubMed.
- F. Chen, E. Zhao, T. Kim, J. Wang, G. Hableel, P. J. T. Reardon, S. J. Ananthakrishna, T. Wang, S. Arconada-Alvarez, J. C. Knowles and J. V. Jokerst, ACS Appl. Mater. Interfaces, 2017, 9, 15566–15576 Search PubMed.
- D. Talwar, N. Poyatos Salguero, C. M. Robertson and J. Xiao, Chemistry, 2014, 20, 245–252 Search PubMed.
- Q. Dou, S. Lei, D.-W. Wang, Q. Zhang, D. Xiao, H. Guo, A. Wang, H. Yang, Y. Li, S. Shi and X. Yan, Energy Environ. Sci., 2018, 11, 3212–3219 Search PubMed.
- H. Huang, Y. Chen, H. Fu, C. Chen, H. Li, Z. Zhang, F. Lai, S. Bai, N. Zhang and T. Liu, J. Energy Chem., 2024, 89, 216–225 Search PubMed.
- N. E. Mendieta-Reyes, W. Cheuquepán, A. Rodes and R. Gómez, ACS Catal., 2019, 10, 103–113 Search PubMed.
- A. T. Ngoc Do, Y. Ha, H. J. Kang, J. M. Kim and J. H. Kwon, J. Hazard. Mater., 2022, 427, 128144 Search PubMed.
- Z. Peng, X. Cao, P. Gao, H. Jia, X. Ren, S. Roy, Z. Li, Y. Zhu, W. Xie, D. Liu, Q. Li, D. Wang, W. Xu and J. G. Zhang, Adv. Funct. Mater., 2020, 30, 2001285 Search PubMed.
- W. Zhang, H. Sun, P. Hu, W. Huang and Q. Zhang, EcoMat, 2021, 3, e12128 Search PubMed.
- P. Garrido-Barros, J. Derosa, M. J. Chalkley and J. C. Peters, Nature, 2022, 609, 71–76 Search PubMed.
- N. Meng, J. Shao, H. Li, Y. Wang, X. Fu, C. Liu, Y. Yu and B. Zhang, Nat. Commun., 2022, 13, 5452 Search PubMed.
- A. Prajapati, B. A. Collins, J. D. Goodpaster and M. R. Singh, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023233118 Search PubMed.
- H. Xu, L. Chen and J. Shi, Energy Environ. Sci., 2023, 16, 1334–1363 Search PubMed.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijoo, P. C. Chen, H. Wang, C. J. Pollock, X. Huang, Y. T. Shao, C. Wang, D. A. Muller, H. D. Abruna and P. Yang, Nature, 2023, 614, 262–269 Search PubMed.
- M. Niermann, A. Beckendorff, M. Kaltschmitt and K. Bonhoff, Int. J. Hydrogen Energy, 2019, 44, 6631–6654 Search PubMed.
- P. Rao and M. Yoon, Energies, 2020, 13, 6040 Search PubMed.
- J. Li, J. Tang, D. Wu, L. Yao and Z. Peng, Int. J. Hydrogen Energy, 2023, 48, 36286–36294 Search PubMed.
- D. Wu, J. Li, L. Yao, R. Xie and Z. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 55292–55298 Search PubMed.
- G. He, H. Liu, X. Liu, Y. Zhu, J. Xiao and L. Han, J. Electroanal. Chem., 2021, 900, 115731 Search PubMed.
- J. Xiao, S. Zhang, Y. Sun, X. Liu, G. He, H. Liu, J. Khan, Y. Zhu, Y. Su, S. Wang and L. Han, Small, 2023, 19, e2206472 Search PubMed.
- X. Qi, K. Obata, Y. Yui, T. Honma, X. Lu, M. Ibe and K. Takanabe, J. Am. Chem. Soc., 2024, 146, 9191–9204 Search PubMed.
- M. Niermann, S. Drünert, M. Kaltschmitt and K. Bonhoff, Energy Environ. Sci., 2019, 12, 290–307 Search PubMed.
- M. Niermann, S. Timmerberg, S. Drünert and M. Kaltschmitt, Renewable Sustainable Energy Rev., 2021, 135, 110171 Search PubMed.
- P. Jena, J. Phys. Chem. Lett., 2011, 2, 206–211 Search PubMed.
- P. M. Modisha, C. N. M. Ouma, R. Garidzirai, P. Wasserscheid and D. Bessarabov, Energy Fuels, 2019, 33, 2778–2796 Search PubMed.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 Search PubMed.
- E. B. A. Tripodi, D. Cespi, F. Passarini, F. Cavani and T. Rossetti, ACS Sustainable Chem. Eng., 2018, 6, 5441–5451 Search PubMed.
- T. Wang, J. Ibañez, K. Wang, L. Fang, M. Sabbe, C. Michel and S. Paul, Nat. Catal, 2019, 2, 773–779 Search PubMed.
- C. Baumler, C. Bauer and R. Kempe, ChemSusChem, 2020, 13, 3110–3114 Search PubMed.
- G. Hahn, P. Kunnas, N. de Jonge and R. Kempe, Nat. Catal., 2018, 2, 71–77 Search PubMed.
- K. Tokmic, B. J. Jackson, A. Salazar, T. J. Woods and A. R. Fout, J. Am. Chem. Soc., 2017, 139, 13554–13561 Search PubMed.
- R. Xia, D. Tian, S. Kattel, B. Hasa, H. Shin, X. Ma, J. G. Chen and F. Jiao, Nat. Commun., 2021, 12, 1949 Search PubMed.
- A. Aguirre and S. E. Collins, Catal. Today, 2019, 336, 22–32 Search PubMed.
- D. Xing, L. Dong, Y. Qi, W. Ge, X. Jiang, Y. Zhao, W. Zhang, P. Tian, H. Jiang and C. Li, AIChE J., 2024, 10, e18377 Search PubMed.
- D. Zhang, J. Chen, Z. Hao, L. Jiao, Q. Ge, W.-F. Fu and X.-J. Lv, Chem. Catal., 2021, 1, 393–406 Search PubMed.
- H. Huang, F. Lai, H. Fu, Y. Chen, H. Li, F. He, Z. Wang, N. Zhang, S. Bai and T. Liu, J. Mater. Chem. A, 2023, 11, 2210–2217 Search PubMed.
- X. Bian, Y. Zou, W. Chen, W. Lin, X. Guo and K. Ding, Appl. Surf. Sci., 2022, 599, 153948 Search PubMed.
- X. Bian, Q. Liu, S. Xie, W. Chen, X. Guo and K. Ding, Appl. Surf. Sci., 2023, 622, 156891 Search PubMed.
- C. Wei, Y. Fang, B. Liu, C. Tang, B. Dong, X. Yin, Z. Bian, Z. Wang, J. Liu, Y. Qian and G. Wang, Nat. Commun., 2023, 14, 3847 Search PubMed.
- K. Müller, Energy Technol., 2022, 10, 2200648 Search PubMed.
- C. M. F. K. A. Pearlstine, J. Am. Chem. Soc., 1986, 108, 5837–5842 Search PubMed.
- G. A. S. Denis and E. Cardin, J. Phys. Chem., 1992, 96, 9424–9431 Search PubMed.
- D. E. G. P. D. Ditlevsen, M. A. Van Hove and G. A. Somorjai, Langmuir, 1993, 9, 1500–1503 Search PubMed.
- I. Waluyo, J. D. Krooswyk, J. Yin, Y. Ren and M. Trenary, ChemCatChem, 2012, 4, 1075–1078 Search PubMed.
- I. Waluyo, J. D. Krooswyk, J. Yin, Y. Ren and M. Trenary, J. Phys. Chem. C, 2013, 117, 4666–4679 Search PubMed.
- E. M. F. Huerta, J. M. Perez, J. L. Vazquez and A. Aldaz, J. Electroanal. Chem., 1999, 469, 159–169 Search PubMed.
- S. E. C. Alejo Aguirre, Catal. Today, 2019, 336, 22–32 Search PubMed.
- D. A. Dobberpuhl and D. C. Johnson, Electroanalysis, 1995, 8, 726–731 Search PubMed.
- W. Ao, H. Ren, C. Cheng, Z. Fan, Q. Qin, P. Yin, Q. Zhang and L. Dai, Angew. Chem., Int. Ed., 2023, 62, e202307924 Search PubMed.
- J. Cho, B. Kim, S. Venkateshalu, D. Y. Chung, K. Lee and S. I. Choi, J. Am. Chem. Soc., 2023, 145, 16951–16965 Search PubMed.
- C. Stumm, M. Kastenmeier, F. Waidhas, M. Bertram, D. J. S. Sandbeck, S. Bochmann, K. J. J. Mayrhofer, J. Bachmann, S. Cherevko, O. Brummel and J. Libuda, Electrochim. Acta, 2021, 389, 138716 Search PubMed.
- M. S. Alias, S. K. Kamarudin, A. M. Zainoodin and M. S. Masdar, Int. J. Hydrogen Energy, 2020, 45, 19620–19641 Search PubMed.
- N. Sazali, W. N. Wan Salleh, A. S. Jamaludin and M. N. Mhd Razali, Membrane, 2020, 10, 99 Search PubMed.
- J. Andersson and S. Grönkvist, Int. J. Hydrogen Energy, 2019, 44, 11901–11919 Search PubMed.
- G. Sievi, D. Geburtig, T. Skeledzic, A. Bösmann, P. Preuster, O. Brummel, F. Waidhas, M. A. Montero, P. Khanipour, I. Katsounaros, J. Libuda, K. J. J. Mayrhofer and P. Wasserscheid, Energy Environ. Sci., 2019, 12, 2305–2314 Search PubMed.
- P. Hauenstein, D. Seeberger, P. Wasserscheid and S. Thiele, Electrochem. Commun., 2020, 118, 106786 Search PubMed.
- M. Brodt, K. Müller, J. Kerres, I. Katsounaros, K. Mayrhofer, P. Preuster, P. Wasserscheid and S. Thiele, Energy Technol., 2021, 9, 2100164 Search PubMed.
- J. O. G. Posada, A. J. R. Rennie, S. P. Villar, V. L. Martins, J. Marinaccio, A. Barnes, C. F. Glover, D. A. Worsley and P. J. Hall, Renewable Sustainable Energy Rev., 2017, 68, 1174–1182 Search PubMed.
- T. Jiang and W. Chen, Curr. Opin. Electrochem., 2021, 30, 100859 Search PubMed.
- S. An, L. Hu, X. Li, S. Zhao, M. Shi and C. Yan, Energy Storage Mater., 2024, 64, 103076 Search PubMed.
- J. Tang, J. Li, P. Pishva, R. Xie and Z. Peng, ACS Energy Lett., 2023, 8, 3727–3732 Search PubMed.
- J. Tang, R. Xie, P. Pishva, X. Shen, Y. Zhu and Z. Peng, J. Mater. Chem. A, 2024, 12, 15580–15591 Search PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Dakota
Braaten
a,
Dakota
Braaten