
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The electrophilic methylating agent [SO2Me]+ parent of two cationic species†
Dirk
Hollenwäger
 *,
Valentin
Bockmair
and
Andreas J.
Kornath‡
*,
Valentin
Bockmair
and
Andreas J.
Kornath‡
Department Chemie Ludwig-Maximilians-Universität München Butenandrstr. 5–13, 81377 München, Germany. E-mail: Dirk.Hollenwaeger@cup.uni-muenchen.de
First published on 27th August 2024
Abstract
Methylfluoride and hydrogen fluoride react with powerful methylating agent [SO2Me]+ in a temperature-dependent addition reaction to form methylated fluorosulfuric acid [FS(OMe)2][Sb2F11] and methylated fluorosulfuric acid methyl ester [FS(OH)(OMe)][SbF6], respectively. The obtained methylated fluorosulfinic acid and methylated fluorosulfinic acid methyl ester were characterized by single X-ray structure analysis and vibrational spectroscopy.
Searching for methylated cations the MeF/Sb5/SO2 system has been investigated many times. The first assumption by Olah in 1969 of the formation of the methyl fluoride antimony pentafluoride complex was disproved by Gillespie in 1971.1,2Gillespie characterized the preparation of an [Me][SbF6] respectively [Me][Sb2F11] salt by 1H NMR spectroscopy.2 In 1975, Peterson invalidated the preparation of the [Me][SbF6] complex by the formation of a methylated sulfur dioxide cation in sulfuryl chloride fluoride.3Peterson’s assumption was supported by new data from Olah in 1975 and Gillespie in the year 1976.3–5Olah confirmed by NMR spectroscopy the formation of the same [SO2Me]+ complex in sulfur dioxide as the solvent with further methylation upon standing to form the [FS(OMe)2]+ cation.5 In 1976, Gillespie published the single crystal X-ray structure of the [SO2Me][Sb2F11] salt.4
We follow up on this early work in the literature by investigating the reaction of the [SO2Me]+ cation in sulfur dioxide with an excess of methyl fluoride and pure anhydrous hydrogen fluoride as the solvent and isolating the resulting cations. Firstly, the starting material [SO2Me][SbF6] was prepared at −60 °C in a FEP vessel tube reactor. The antimony pentafluoride was condensed into the FEP reactor and sulfur dioxide as reagent and solvent was added, according to eqn (1). Methyl fluoride was added to the reaction and warmed up to −60 °C. The equivalents of 1.0 to 2.0 SbF5 to 0.9 to 1.0 MeF were tested. The solvent was removed overnight at −78 °C. The [FS(OMe)2][Sb2F11] salt was obtained by dissolving [SO2Me][SbF6] in sulfur dioxide and with 1.0 to 10.0 equivalents of methyl fluoride. The mixture was warmed up in 10 °C steps from −70 °C to room temperature. Both reactions were traced by 1H and 19F-NMR. After warming up to room temperature in both reactions, the solvent was removed at −78 °C overnight. The temperature-dependent addition reaction of methyl fluoride to the [SO2Me]+ cation can be traced by 19F NMR spectroscopy in 10 °C steps from −70 °C to room temperature. Fig. 1 shows the stacked measured spectra of [SO2Me]+ cation in SO2 with a methyl fluoride excess between 0 to +80 ppm. The 19F NMR measurements show, that at the beginning two species are observed, the [FS(OMe)O]·SbF5 adduct and the [FS(OMe)2]+ cation. The formation of the [FS(OMe)2]+ cation takes place as soon as the solvent SO2 becomes liquid. This reaction is very slow at −70 °C, so that the formation, of the [FS(OMe)O]·SbF5 can be traced. The temperature-dependent NMR scan shows, that the percentage of [FS(OMe)O]·SbF5 (s, 27.56 ppm at −70°) decreases with every temperature step, and the proportion of the [FS(OMe)2]+ cation (s, 16.18 ppm at −70°) increases. At −20 °C the formation of [FS(OMe)2]+ is fast enough, that only traces of the adduct can be measured. Eqn (1) shows the assumed reaction.
 | (1) |
 | ||
| Fig. 1 19F NMR spectra of [FS(OMe)2][Sb2F11] from −70 °C to 26 °C in SO2. | ||
Fig. 2 displays the low-temperature Raman and room-temperature IR spectra of [FS(OMe)2][Sb2F11]. The Supporting Information shows in Table S1 (ESI†) a complete list of the experimental data listed together with the quantum chemically calculated frequencies of the [FS(OMe)2]+ cation. The cation has a C1 symmetry with 30 fundamental vibrational modes. The Raman spectra detected three C–H stretching vibrations at 2855 cm−1, 2994 cm−1, and 3088 cm−1. In the literature, only one stretching vibration at 2985 cm−1 is reported for the methylated SO2.4 In the IR spectra the C–H stretching vibrations are detected at 2987 cm−1 and 3094 cm−1. The S–O stretching vibration is red-shifted in the Raman spectrum compared to the starting material approximately by 307 cm−1 to 1008 cm−1, respectively 986 cm−1.4 In the starting material the S–O stretching vibration is reported in the Raman spectrum at 1315 cm−1, as well as 995 cm−1.4 Both stretching vibrations are IR active and are observed at 1022 cm−1, as well as 960 cm−1. Another indication for the methyl fluoride addition is the C–F stretching vibration observed at 813 cm−1 in Raman, respectively 808 cm−1 in IR spectroscopy. Characteristic bands for the [Sb2F11]− anion are also observed.
 | ||
| Fig. 2 Low-temperature Raman and room temperature IR of [FS(OMe)2][Sb2F11]. | ||
The crystal structure of [FS(OMe)2][Sb2F11] crystallizes in the monoclinic space group P21/n with four formula units per unit cell. The Supporting Infomation displays the crystallographic data in Table S3 (ESI†), as well as a complete list of the bond length, angles, and donor–acceptor interactions (Table S4, ESI†). Fig. 3 shows the asymmetric unit of [FS(OMe)2][Sb2F11]. The S1–O1 bond length is determined to be 1.517(6) Å, respectively the S1–O2 bond length is measured to be 1.545(5) Å and comparable to the 1.491(10) Å bond length reported in the literature.4 Compared to the S–O double bond (1.379(10) Å) in the [SO2Me]+ the S–O bonds are significantly elongated.4 The S1–F1 bond length (1.558(5) Å) is in the same area as in the [FS(OH)2]+ cation (1.547(7) Å).6 The C1–O1 (1.476(9) Å) and C1–O2 (1.488(9) Å) are in the same range than in methylated SO2 (1.492(17) Å).4 The [Sb2F11]− anion displays Sb–F with bond lengths of 1.849(4) Å and 2.031(4) Å for terminal and bridging Sb–F bonds, respectively. These bond lengths are comparable to values reported in the literature for [Sb2F11]− anions.7,8 The crystal structure is characterized by three S–F interactions, which are significant below the sum of the van der Waals radii (3.27 Å) by 89% (S1−F2i; 2.996(5) Å), 92% (S1–F3; 2.910(4) Å) and 94% (S1–F12iv; 3.066(5) Å).9
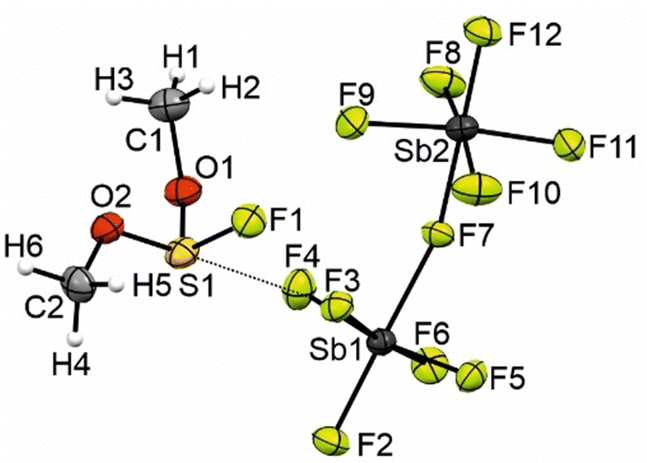 | ||
| Fig. 3 Asymmetric unit of [FS(OMe)2][ Sb2F11]. (displacement ellipsoids with 50% probability). | ||
If [SO2Me]+ is reacted with an excess of CH3F in HF instead of SO2, the room-temperature stable [FS(OH)(OMe)]+ cation is formed. In contrast to the protonated species [FS(OH)2][SbF6], the salts of [FS(OMe)2][Sb2F11] and [FS(OH)(OMe)][SbF6] are stable at room temperature.6 The assumed reaction is shown in eqn (2). The reaction is also scanned in 10° steps in NMR spectroscopy. Fig. 4 shows the 19F NMR spectroscopy measurements of the formation of the [FS(OH)(OMe)]+ cation. Three compounds are identified in the 19F NMR spectrum, the [FS(OMe)O]·SbF5 adduct (s, 28.50 ppm at −70°), [FS(OMe)2]+ (s, 13.28 ppm at −70°) as well as the [FS(OH)(OMe)]+ cation (s, 66.15 ppm at −70°). The [FS(OMe)2]+ cation is the proportion of [FS(OMe)2]+ decreases with every temperature step. At −20 °C the formation of [FS(OH)(OMe)]+ is favoured, and only traces of the SbF5 adduct are measured. The equilibrium of methylated SO2 and the [FS(OMe)O]·SbF5 adduct decreases very rapidly after the temperature step from −20 °C to −10 °C. The main compound at −10 °C is the desired [FS(OH)(OMe)]+ cation. From −70 °C to −20 °C the formation of the [FS(OH)(OMe)]+ cation is kinetically slowed down by temperature.
 | (2) |
 | ||
| Fig. 4 19F NMR spectra of [FS(OH)(OMe)][SbF6] from −70 °C to 25 °C in aHF. | ||
Fig. 5 displays the room-temperature IR and low-temperature Raman spectra of [FS(OH)(OMe)]+. The Supporting Information is shown in Table S2 (ESI†) a complete list of the experimental data listed together with the quantum chemically calculated frequencies of the [FS(OH)(OMe)]+·HF cation. The cation has a C1 symmetry with 21 fundamental vibrational modes. The Raman spectra detected two C–H stretching vibrations at 2986 cm−1 and 3085 cm−1. In the literature is only one stretching vibration at 2985 cm−1 reported for the methylated SO2.4 In IR spectra one C–H stretching vibration is detected at 3111 cm−1. In IR spectra the O–H stretching vibration is detected at 3284 cm−1. The S–O stretching vibration is red-shifted compared to the starting material (1315 cm−1, respectively 995 cm−1) approximately by 317 cm−1 to 998 cm−1.4 Both stretching vibrations are IR active and are observed at 1029 cm−1, as well as 959 cm−1. The C–F stretching vibration is observed at 835 cm−1 in IR spectroscopy.
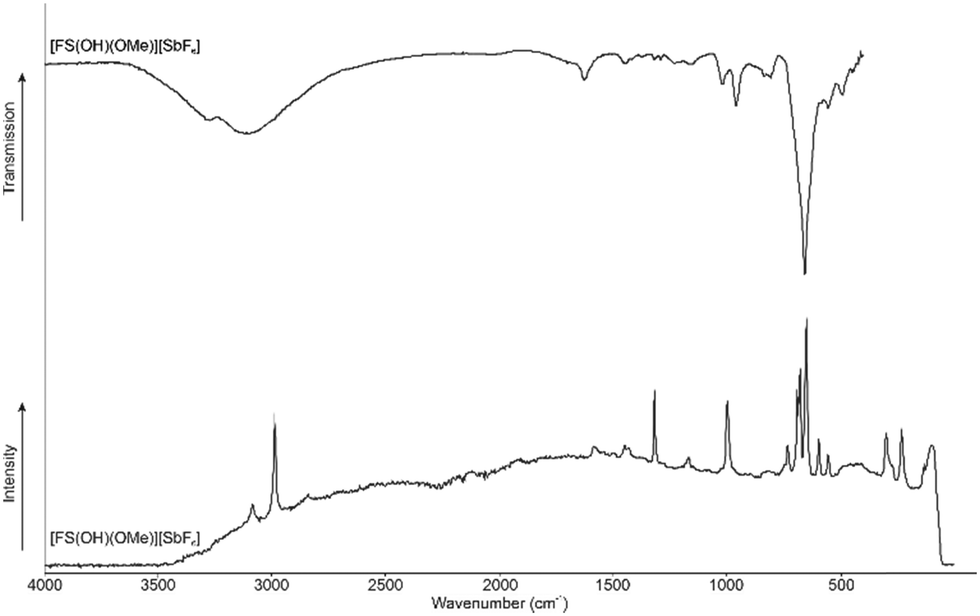 | ||
| Fig. 5 Low-temperature Raman and room temperature IR of [FS(OH)(OMe)][SbF6]. | ||
The crystal structure of [FS(OH)(OMe)][SbF6] crystallizes in the orthorombic space group Pca21 with four formula units per unit cell. The Supporting Infomation displays the crystallographic data in Table S3 (ESI†), as well as a complete list of the bond length, angles, and donor–acceptor interactions (Table S5, ESI†). In Fig. 3 is shown the asymmetric unit of [FS(OH)(OMe)][SbF6]. The S–O bond lengths of the protonated side are in the range between S3–O5 1.521(7) Å to S1–O1 1.532(6) Å and are in the same range as in [FS(OMe)2]+ (1.522(8) Å and 1.537(7) Å).6 The S–O bond lengths at the methylated side are in the range between S1–O2 1.507(6) Å to S4–O8 1.517(6) Å and slightly elongated compared to the [SO2Me]+ (1.492(17) Å). The C–O bond lengths are between C3–O6 1.484(12) Å and C4–O8 1.496(11) Å and in the same range compared to methylated SO2 (1.492(17) Å). The S–F bond lengths are between S1–F1 1.549(5) Å and S2–F2 1.561(6) Å and in the range of the S–F bond lengths of [FS(OH)2]+ (1.547(7) Å). The bond lengths of the [SbF6]− anion are in the range of values reported in the literature.10–12 The S1–F14 (2.743(6) Å) and S1–F6 (2.751(5) Å) distances are 84% below of the sum of the van der Waals radii (3.27 Å) (Fig. 6).9
 | ||
| Fig. 6 Asymmetric unit of [FS(OH)(OMe)][SbF6]. (displacement ellipsoids with 50% probability). | ||
The quantum chemical calculations were performed at the M06-2X/aug-cc-pVTZ level of theory with the Gaussian16 program package.13 The internal reaction coordinate (IRC) calculations of the 1,2-addition of an HF molecule to the [SO2Me]+ are shown in Fig. 7. The minimal potential energy curve has been calculated along the intrinsic reaction coordinate of the addition reaction from the transition state. The optimized structures are calculated with implicit water solvation. It has been reported in the literature that protic solvents can lower the calculated intrinsic barriers compared to the gas phase and facilitate ion transfer.14–16 Water is used because it has similar properties compared to anhydrous HF. The use of a protic solvent, which is also capable of autoprotolysis is important for more precise calculations. In the quantum chemical calculation, an exothermal reaction is predicted, which gains 35.6 kJ mol−1 by the addition of the HF molecule to the [SO2Me]+ cation. The reaction has a high energy barrier of 147.7 kJ mol−1. Nevertheless, the reaction is very likely to take place under these conditions, as it proceeds slowly at low temperatures and accelerates as the temperature increases. This is consistent with the complete shift to the product at higher temperatures in NMR spectroscopy, as the reaction is irreversible.
 | ||
| Fig. 7 IRC calculations of the HF addition to the [SO2Me]+ cation at M062X/aug-cc-pVTZ level of theory. | ||
In Fig. S11 in the ESI† are shown the Mapped Electrostatic Potentials (MEP). The MEPs were calculated together with Natural Population Analysis (NPA) charges to investigate the 1,2-addition of HF to the [SO2Me]+ cation. According to the calculations, the most negative electrostatic potential (red) is located at the not methylated oxygen in the starting material. The fact that protons are oriented towards negative electrostatic potentials of oxygen is reported in the literature for a large number of protonations.17–19 The most positive potential (blue) is located at the sulphur. The starting material [SO2Me]+ therefore has a π hole on the sulphur, so that the fluoride can easily attach to it.20–22
In conclusion, we have successfully isolated the salts [FS(OMe)2][Sb2F11] and [FS(OH)(OMe)][SbF6]. The salts were characterized by NMR spectroscopy and for the first time by vibrational spectroscopy and single-crystal X-ray diffraction. The crystal structures allow a better understanding of the reaction, which has been described and discussed controversially in the literature. The [FS(OH)(OMe)][SbF6] crystal structure is characterized by two S–F interaction of 84% of the van der Waals radii. The crystal structure of [FS(OMe)2][Sb2F11] has three S–F interaction between 89% and 94% of the van der Waals radii. The quantum mechanical calculations at the M062X-aug-cc-pVTZ level of theory support the experimental data of an exothermic reaction. The experimental data and quantum chemical calculations show the importance of low temperature in syntheses using the SO2/MeF/SbF5 methylation reagent.
Data availability
The data supporting this article have been included as part of the ESI.† For full details on vibrational spectroscopy, NMR spectroscopy, X-ray diffraction refinement, and computational details. Crystallographic data has been deposited at the CCDC under CCDC 2368718 and 2368721 can be obtained from https://www.ccdc.cam.ac.uk.Conflicts of interest
There are no conflicts to declare.References
- G. A. Olah, J. R. DeMember and R. H. Schlosberg, J. Am. Chem. Soc., 1969, 91, 2112 CrossRef CAS.
- J. Bacon and R. J. Gillespie, J. Am. Chem. Soc., 1971, 93, 6914 CrossRef CAS.
- P. E. Peterson, R. Brockington and D. W. Vidrine, J. Am. Chem. Soc., 1976, 98, 2660 CrossRef CAS.
- R. J. Gillespie, F. G. Riddell and D. R. Slim, J. Am. Chem. Soc., 1976, 98, 8069 CrossRef CAS.
- G. A. Olah, D. J. Donovan and H. C. Lin, J. Am. Chem. Soc., 1976, 98, 2661 CrossRef CAS.
- A. Kornath, R. Seelbinder and R. Minkwitz, Angew. Chem., Int. Ed., 2005, 44, 973–975 CrossRef CAS.
- R. Minkwitz and S. Reinemann, Z. anorg. allg. Chem., 1999, 625, 121–125 CrossRef CAS.
- M. D. Lind and K. O. Christe, Inorg. Chem., 1972, 11, 608–612 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. Minkwitz, R. Lekies and H. Preut, Z. Naturforsch. B, 1987, 42, 1227–1230 CrossRef CAS.
- R. Minkwitz, C. Hirsch and T. Berends, Eur. J. Inorg. Chem., 1999, 2249–2254 CrossRef CAS.
- R. Minkwitz and S. Schneider, Angew. Chem., Int. Ed., 1999, 38, 210–212 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel and G. E. Scuseriaet al., Gaussian, C.01, Wallingford, CT, 2016 Search PubMed.
- W. Chansen and N. Kungwan, J. Phys. Chem., 2021, 125, 5314–5325 CrossRef CAS.
- B. Xiao, Y. Li, X. Yu and J. Cheng, J. Phys. Chem., 2015, 119, 11882–11890 CrossRef CAS.
- H. Tavakol, J. Phys. Chem., 2013, 117, 6809–6816 CrossRef CAS.
- S. Steiner, A. Nitzer, C. Jessen and A. J. Kornath, Z. anorg. allg. Chem., 2024, 650 DOI:10.1002/zaac.202400013.
- S. Steiner, C. Jessen and A. J. Kornath, Z. anorg. allg. Chem., 2022, 648 DOI:10.1002/zaac.202200060.
- M. Schickinger, T. Saal, F. Zischka, J. Axhausen, K. Stierstorfer, Y. Morgenstern and A. J. Kornath, ChemistrySelect, 2018, 3, 12396–12404 CrossRef CAS.
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS.
- G. A. Olah, G. Liang and Y. K. Mo, J. Org. Chem., 1974, 39, 2394–2398 CrossRef CAS.
- G. Frenking, W. Koch and H. Schwarz, J. Comput. Chem., 1986, 7, 406–416 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2368718 and 2368721. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03400g |
| ‡ Prof. Dr A.J. Kornath passed away unexpectedly in March 2024. |
| This journal is © The Royal Society of Chemistry 2024 |