
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Orally bioavailable STING antagonist synthesized via multi-component Povarov–Doebner type reaction†
Kofi B.
Owusu
ab,
Jyotrimayee
Samal
ab,
Delmis E.
Hernandez
a and
Herman O.
Sintim
 *abc
*abc
aDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, USA. E-mail: hsintim@purdue.edu
bInstitute for Drug Discovery, Purdue University, 720 Clinic Drive, West Lafayette, Indiana 47907, USA
cPurdue Institute of Inflammation, Immunology, and Infectious Disease, West Lafayette, IN 47907, USA
First published on 20th September 2024
Abstract
Aberrant activation of the cGAS-STING signaling results in innate immune response induction. Herein, we report HSKB142, an orally bioavailable compound containing the 3H-pyrazolo [4,3-f]quinoline synthesized via a Povarov–Doebner MCR. HSKB142 is non-cytotoxic towards immune cells and suppresses type-1 interferon expression in human THP-1 monocytes upon treatment with 2′3′-cGAMP.
The stimulator of interferon genes, STING has gained widespread attention since the seminal discovery by Barber and Ishikawa in 2008.1 STING is now appreciated as a key regulator of chronic inflammation, metabolic diseases as well as cancer.2,3 As an important protein that plays a critical role in the immune system's response to infection, the cGAS-STING pathway is essential for detecting and limiting the transmission of extraneous DNA.4,5 The accumulation of double stranded DNA (dsDNA) in the cytosol other than in the nucleus or mitochondria of the cell, where it is natively bound, suggests the possibility of infection, uptake of exogeneous dsDNA, or lack of activity of nucleases such as TREX1 and DNAse II.1,5,6 As a defensive mechanism, mammals have developed a host of mechanisms, such as involving TLR9,7 ZBP1,8 AIM2,9 IFI16,10 through which dsDNA is detected with the prominent amongst these being the cGMP-AMP synthase (cGAS),11,12 a cytosolic nucleotidyltransferase. Upon sensing misplaced genomic, mitochondrial, and microbial double-stranded DNA (dsDNA), cGAS dimerizes and forms a complex of two cGAS molecules and two dsDNA molecules. Further conformational changes allow cGAS to catalyze the coupling of ATP to GTP and cyclization to generate the second messenger, cyclic guanosine monophosphate-adenosine monophosphate, cGAMP.13 cGAMP diffuses through the cytosol to bind to the universal cyclic dinucleotide sensor protein, STING, a protein localized to the endoplasmic reticulum (ER). After cGAMP binding, STING undergoes a conformation change that promotes oligomerization and subsequent translocation from the ER to the Golgi apparatus leading to its recruitment and further phosphorylation of tank binding kinase I (TBK1).14 Phosphorylated TBK1 then recruits and phosphorylates interferon regulatory factor 3 (IRF3) and nuclear factor kappa-light-chain- enhancer of activated B cells (NFκB) leading to their trafficking into the nucleus where these proteins bind DNA and activate certain cytokines, including the transcription of type I interferons (IFN), which are essential towards mounting appropriate immune responses against pathogen invasion.15–17
Through the activation of the cGAS-STING pathway, the innate immune defense is beneficial in mammals but the aberrant activation of this axis leads to robust inflammatory response, which can lead to the development of chronic autoimmune disorders and severe pathology in human diseases such as amyotrophic lateral sclerosis (ALS),18 familial chilblain lupus,19 Aicardi-Goutieres syndrome (AGS),20–22 Lupus,23 and STING associated vasculopathy with onset in infancy (SAVI).24,25
Recent studies have shown that the cGAS-STING pathway plays an important role in acute kidney injury (AKI), a disease characterized by a rapid decline in renal function with the possibility of chronic kidney disease (CKD) setting in.26–30 With such a central role played by activated cGAS-STING axis in numerous diseases, efforts have begun to intensify in the identification and development of STING inhibitors (see Fig. 1 for examples of STING antagonists).31–36
 | ||
| Fig. 1 Structures of reported small molecule STING antagonists. | ||
Our group previously reported novel class of STING antagonists, such as HSD1077 (Fig. 1), which are synthesized via multi-component Povarov–Doebner type reaction.31 Using a fluorescence polarization assay (Fig. 2), developed in our group,37 we demonstrated that HSD1077 could compete with fluorescein-labeled c-di-GMP in binding hSTING CTD containing residues 155–341, which has previously been shown to be soluble and capable of binding to CDNs in solution.38 At 100 nM, HSD1077 was able to inhibit IL-6 production in both THP-1 and Raw macrophage cell lines. Mechanistic studies revealed that HSD1077 inhibited both TBK1 and STING phosphorylation.31 While HSD1077 is orally bioavailable [(F = 91%)], unfortunately it suffers from high clearance (Clobs = 222 mL min−1 kg−1 after IV dosing of 2 mg Kg−1) in mice.
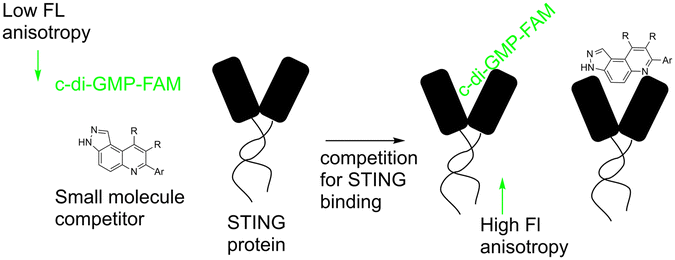 | ||
| Fig. 2 Schematic diagram showing competitive binding of F-c-di-GMP in STING by a small molecule. | ||
We were therefore motivated to continue searching for STING antagonists that could be used in vivo. Our group has identified many compounds that contain the same 3H-pyrazolo[4,3-f]quinoline scaffold in HSD1077, which are efficacious in vivo.39 Therefore, we reasoned that the unsubstituted pyrazole group in HSD1077 might be responsible for the PK liability. Unfortunately, substituting the pyrazole in HSD1077 to potentially reduce metabolic inactivation also reduced STING binding. Luckily, screening our proprietary 3H-pyrazolo[4,3-f]quinoline-containing library identified benzamides 1 and 2 (see Fig. 3) as potential STING binders. Herein, we detail structure–activity relationship studies of these new STING binders. Pleasingly HSKB143 (compound class A, Fig. 3) and HSKB142 (compound class B, Fig. 3) antagonize STING. However, only HSKB142 is bioavailable while HSKB143 is not bioavailable when dosed orally.
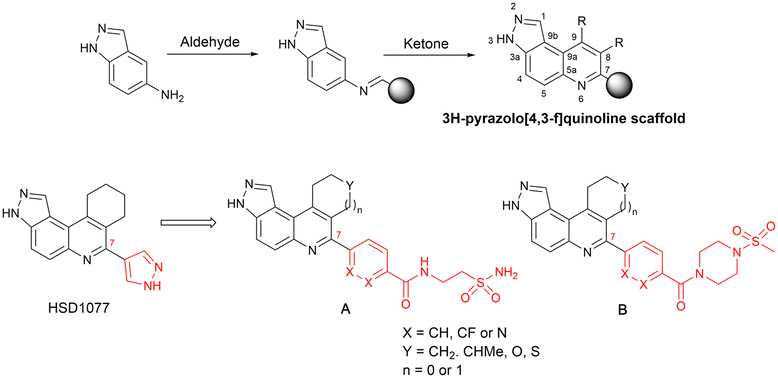 | ||
| Fig. 3 Synthesis of quinoline compounds using the Povarov–Doebner type multicomponent reaction. STING binders, HSD1077, compound of types A and B. | ||
We synthesized 22 compounds via a tandem one-pot three-component Doebner/Povarov type MCR followed by amidation (Fig. 4). With potential STING binders in hand (Fig. 5), we proceeded to evaluate binding to STING, using the binding assay described in Fig. 2.
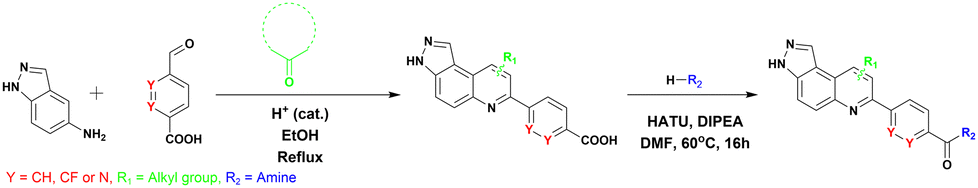 | ||
| Fig. 4 Synthetic steps for synthesis of analogues. | ||
 | ||
| Fig. 5 Compounds screened for STING binding. | ||
Briefly, 20 μM of compounds and 50 nM of the c-di-GMP-fluorescein probe (c-di-GMP-FAM) were incubated with 10 μM of hSTING for 5 minutes prior to evaluation of fluorescence anisotropy. As a positive control, we used ADU-S100, a known agonist of STING that binds to the cyclic dinucleotide binding site of STING.40 It was observed that compounds containing ethyl piperazine 1, morpholine 2, methyl piperidinol 3, thiomorpholine dioxide 4, and dimethyl morpholine 5 benzamides were poor STING binders, whereas methylsulfonyl piperazine 6 could bind to STING, albeit only moderately (Table 1 and Fig. S1, ESI†). Further substitutions of methylsulfonyl piperazine analog 6 revealed that fluorine substitution on the benzamide moiety, 7 and HSKB142, improved STING binding whereas changing the benzamide to picolinamide 8 or nicotinamide 9 abrogated STING binding (Table 1). Substituting the ring appended to positions 8 and 9 of pyrazolo[4,3-f]quinoline core with OH (10, 13) or Me (11), and replacing a carbon with S (12) did not improve STING binding. Compound 14, the tetrahydro-2H-pyran analog of compound 6 was equally good at binding to STING. The N-(2-sulfamoylethyl)benzamide analogs, HSKB143, 17–19 could also bind to STING. As expected, the nicotinamide or picolinamide analogs, 16 and 17, were not STING binders (Table 1).
With HSKB142 and 143 as lead compounds from the single concentration screen, we proceeded to determine the half maximal inhibitory concentration (IC50) values for competing with c-di-GMP-FAM binding to STING. HSKB143 and HSKB142 could compete with c-di-GMP-FAM with IC50 of 10 and 12 μM respectively (Fig. 6A). To validate the c-di-GMP-FAM binding assay, we employed differential scanning fluorimetry (DSF) assay to measure the shifts in temperature which arises upon stabilization of hSTING41 when HSKB142, HSKB143, HSD1077 are bound. We used H151,32 a known STING antagonist that binds to a non-CDN site and cGAMP, the native ligand, as controls. HSKB142, HSD1077, HSKB143 caused ΔTm = 4 °C, 3 °C and 2 °C respectively, whereas H151 caused ΔTm = 1 °C. These values are significantly less than the thermal shift for 2′3′-cGAMP (ΔTm = 15 °C) but nonetheless show that the antagonists do bind to STING and cause protein stabilization.
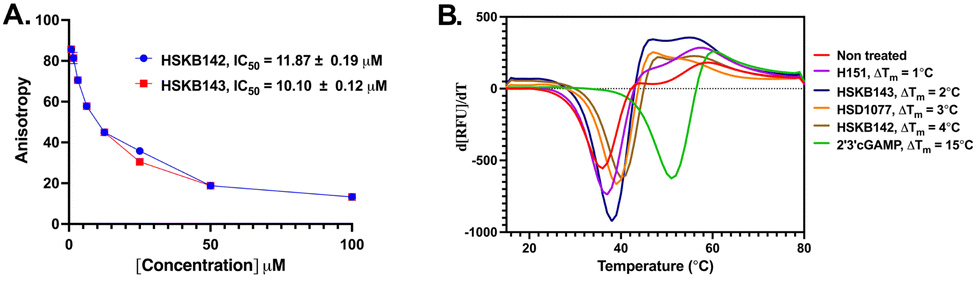 | ||
| Fig. 6 Biochemical assays to characterize STING. (A) IC50 curves of inhibitors displacing c-di-GMP-FAM from STING. (B) Second derivative of the thermal shift assay varying temperature from 15 °C to 80 °C with ΔTm values. | ||
Next, we investigated whether HSKB142 and HSKB143 could attenuate the expression of interferons in human derived THP-1 monocytes via the STING pathway upon cGAMP stimulation. Using THP-1 dual cells incorporated with a luciferase gene under the control of an ISG54 inducible promoter, relative quantifications of type 1 interferon expression could be achieved through luciferase detection upon cGAS–STING pathway induction. Pre -treating THP-1 monocytes with HSKB142, HSKB143 and H151 (known Sting antagonist)32 reduced cGAMP activated type 1 interferon expression in a dose dependent manner with IC50 values of 8, 10 and 29 nM respectively. Similarly, the compounds could antagonize the action of cGAMP in THP-1 dual cells expressing a knock in constitutionally active STING (N154S), with IC50 of 25 nM (HSKB142), 28 nM (HSKB143) and 39 nM (H151), Fig. 7B.
 | ||
| Fig. 7 HSKB142 and HSKB143 attenuate interferon induction in human THP-1 cells. (A) Treatment of HSKB142 and HSKB143 to THP-1 dual monocytes. (B) Treatment of HSKB142 and HSKB143 to THP-1 dual (KI STING N154S). | ||
HSKB142 and HSKB143 were not cytotoxic when tested against immune cells, Jurkat, RAW and THP-1 macrophages at a higher concentration of 10 μM (Fig. S2, ESI†). For successful clinical translation of STING antagonists, it is essential that the compounds are bioavailable. So, we evaluated the pharmacokinetic properties of HSKB142 and 143 after dosing at 20 mg kg−1via oral gavage (PO). As shown in (Fig. S5B, ESI†), the plasma concentration of HSKB142 peaked at 0.5 hours after oral administration (Cmax = 1650 ng mL−1) and dropped to around 6.73 ng mL−1 after 24 hours. HSKB143, on the other hand, displayed poor oral bioavailability with Cmax = 4.5 ng mL−1. We speculate that the poor oral availability of HSK143 is probably due to hydrolysis of the amide bond. Comparing the pharmacokinetic parameters of HSKB142 to the earlier reported HSD1077 (see Fig. 8 and Tables S1–S4, ESI†), HSKB142 shows enhanced properties such as a lower clearance, Clobs, of 32.7 mL min−1 kg−1 after IV dosing of 2 mg kg−1 compared to Clobs of 222 mL min−1 kg−1 after IV dosing of 2 mg kg−1 for HSD1077. The exposure of HSKB142 (AUClast = 4147.6 h ng mL−1 after PO dosing of 20 mg kg−1) is also higher than that of HSD1077 (AUClast = 1272.0 h ng mL−1 after PO dosing of 20 mg kg−1).
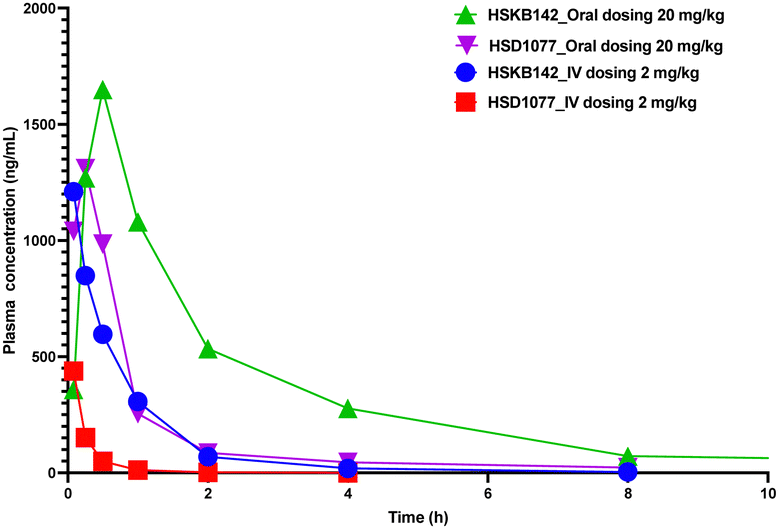 | ||
| Fig. 8 Total plasma concentration vs. time profile for HSKB142 and HSD1077 after 20 mg kg−1 dosing (PO) and 2 mg kg−1 dosing (IV) in Male CD1 Mice (n = 3). The three samples were pooled together, and drug concentration analyzed via LC-MS (as contract research at Pharmaron). | ||
In conclusion, using the Povarov–Doebner type reaction, we have developed 3H-pyrazolo[4,3-f]quinoline containing compounds that inhibit the cGAS–STING pathway. HSKB142 is orally bioavailable and serves as a lead compound for the development of novel therapeutics targeting STING for the potential treatment of chronic autoimmune diseases, such as SAVI and AGS.
We thank Purdue University for funding.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- H. Ishikawa and G. N. Barber, Nature, 2008, 455, 674–678 CrossRef CAS PubMed.
- J. Gong, X. Gao, S. Ge, H. Li, R. Wang and L. Zhao, Int. J. Biol. Sci., 2024, 20, 152–174 CrossRef CAS PubMed.
- G. N. Barber, Nat. Rev. Immunol., 2015, 15, 760–770 CrossRef CAS PubMed.
- J. M. Jenson, T. Li, F. Du, C. K. Ea and Z. J. Chen, Nature, 2023, 616, 1–3 CrossRef PubMed.
- H. Ishikawa, Z. Ma and G. N. Barber, Nature, 2009, 461, 788–792 CrossRef CAS PubMed.
- A. Ablasser and Z. J. Chen, Science, 2019, 363, 8657 CrossRef PubMed.
- H. Hemmi, O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda and S. Akira, Nature, 2000, 408, 740–745 CrossRef CAS PubMed.
- A. Takaoka, Z. Wang, M. Choi, H. Yanai, H. Negeshi, T. Ban, Y. Lu, M. Miyagashi., T. Kodama, K. Honda, Y. Ohba and T. Taniguchi, Nature, 2007, 448, 501–505 CrossRef CAS PubMed.
- T. Bürckstümmer, C. Baumann, S. Blüml, E. Dixit, G. Dürnberger, H. Jahn, M. Planyavsky, M. Bilban, J. Colinge, K. L. Bennett and G. Superti-Furga, Nat. Immunol., 2009, 10, 266–272 CrossRef PubMed.
- L. Unterholzner, S. Keating, M. Baran, K. A. Horan, S. O. Jensen, S. Sharma, C. M. Sirois, T. Jin, E. Latz, T. S. Xiao, K. A. Fritzgerald, S. O. Paludan and A. G. Bowie, Nat. Immunol., 2010, 11, 997–1004 CrossRef CAS PubMed.
- J. Wu, L. Sun, X. Chen, F. Du, H. Shi, C. Chen and Z. J. Chen, Science, 2012, 339, 826–830 CrossRef PubMed.
- L. Sun, J. Wu, F. Du, X. Chen and Z. J. Chen, Science, 2013, 339, 786–791 CrossRef CAS PubMed.
- C. Chen and P. Xu, Trends Cell Biol., 2023, 33, 630–648 CrossRef CAS PubMed.
- G. Shang, C. Zhang, Z. J. Chen, X. Bai and X. Zhang, Nature, 2019, 567, 389–393 CrossRef CAS PubMed.
- X. Gui, H. Yang, T. Li, X. Tan, P. Shi, M. Li, F. Du and Z. J. Chen, Nature, 2019, 567, 262–266 CrossRef CAS PubMed.
- S. Yum, M. Li, Y. Fang and Z. J. Chen, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(14), e2100225118 CrossRef CAS PubMed.
- D. Liu, H. Wu, C. Wang, Y. Li, H. Tian, S. Siraj, S. Sehgal, X. Wang, J. Wang, Y. Shang, Z. Jiang, L. Liu and Q. Chen, Cell Death Differ., 2019, 26, 1735–1749 CrossRef CAS PubMed.
- C. K. Glass, K. Saijo, B. Winner, M. C. Marchetto and F. H. Gage, Cell, 2010, 140, 918–934 CrossRef CAS PubMed.
- N. König, C. Fiehn, C. Wolf, M. Schuster, E. Cura Costa, V. Tüngler, H. A. Alvarezo., O. Chara, K. Engel, R. Goldbach-Mansky, C. Günther and M. A. Lee-Kirsch, Ann. Rheum. Dis., 2017, 76, 468–472 CrossRef PubMed.
- A. Decout, J. D. Katz, S. Venkatraman and A. Ablasser, Nat. Rev. Immunol., 2021, 21, 548–569 CrossRef CAS PubMed.
- Y. J. Crow and N. Manel, Nat. Rev. Immunol., 2015, 15, 429–440 CrossRef CAS PubMed.
- V. Pokatayev, N. Hasin, H. Chon, S. M. Cerritelli, K. Sakhuja, J. M. Ward, H. D. Morris, N. Yan and R. J. Crouch, J. Exp. Med., 2016, 213, 329–336 CrossRef CAS PubMed.
- Y. Kato, J. Park, H. Takamatsu, H. Konaka, W. Aoki, S. Aburaya, M. Ueda, M. Nishide, S. Koyama, Y. Hayama, Y. Kinehara, T. Hirano, Y. Shima, M. Narazaki and A. Kumanogoh, Ann. Rheum. Dis., 2018, 77, 1507–1515 CrossRef CAS PubMed.
- S. Skopelja-Gardner, J. An and K. B. Elkon, Nat. Rev. Nephrol., 2022, 18, 558–572 CrossRef CAS PubMed.
- N. Jeremiah, B. Neven, M. Gentili, I. Callebaut, S. Maschalidi, M. C. Stolzenberg, N. Goudin, M. L. Frémond, P. Nitschke, T. J. Molina, S. Blanche, C. Picard, G. I. Rice, Y. J. Crow, N. Manel, A. Fischer, B. Bader-Meunier and F. Rieux-Laucat, J. Clin. Invest., 2014, 124, 5516–5520 CrossRef PubMed.
- C. Sun, H. Shi, X. Zhao, Y. Chang, X. Wang, S. Zhu and S. Sun, J. Inflammation Res., 2023, 16, 4461–4470 CrossRef CAS PubMed.
- S. R. Mulay, A. Linkermann and H. J. Anders, J. Am. Soc. Nephrol., 2016, 27, 27–39 CrossRef CAS PubMed.
- M. Zhao, Y. Wang, L. Li, S. Liu, C. Wang, Y. Yuan, G. Yang, Y. Chen, J. Cheng, Y. Lu and J. Liu, Theranostics, 2021, 11, 1845–1863 CrossRef CAS PubMed.
- L. S. Huang, Z. Hong, W. Wu, S. Xiong, M. Zhong, X. Gao, J. Rehman and A. B. Malik, Immunity, 2020, 52, 475–486 CrossRef CAS PubMed.
- H. Maekawa, T. Inoue, H. Ouchi, T. Jao, R. Inoue, H. Nishi, R. Fujii, F. Ishidate, T. Tanaka, Y. Tanaka, N. Hirokawa, M. Nangaku and R. Inagi, Cell Rep., 2019, 29, 1261–1273 CrossRef CAS PubMed.
- W. W. S. Ong, N. Dayal, R. Chaudhuri, J. Lamptey and H. O. Sintim, RSC Med. Chem., 2023, 14, 1101 RSC.
- S. M. Haag, M. F. Gulen, L. Reymond, A. Gibelin, L. Abrami, A. Decout, M. Heymann, F. G. Van Der Goot, G. Turcatti, R. Behrendt and A. Ablasser, Nature, 2018, 559, 269–273 CrossRef CAS PubMed.
- S. Li, Z. Hong, Z. Wang, F. Li, J. Mei, L. Huang, X. Lou, S. Zhao, L. Song, W. Chen, Q. Wang, H. Liu, Y. Cai, H. Yu, H. Xu, G. Zeng, Q. Wang, J. Zhu, X. Liu, N. Tan and C. Wang, Cell Rep., 2018, 25, 3405–3421 CrossRef CAS PubMed.
- T. Siu, M. D. Altman, G. A. Baltus, M. Childers, J. M. Ellis, H. Gunaydin, H. Hatch, T. Ho, J. Jewell, B. M. Lacey, C. A. Lesburg, B. S. Pan, B. Sauvagnat, G. K. Schroeder and S. Xu, ACS Med. Chem. Lett., 2019, 10, 92–97 CrossRef CAS PubMed.
- L. Barasa, S. Chaudhuri, J. Y. Zhou, Z. Jiang, S. Choudhary, R. M. Green, E. Wiggin, M. Cameron, F. Humphries, K. A. Fitzgerald and P. R. Thompson, J. Am. Chem. Soc., 2023, 145, 20273–20288 CrossRef CAS PubMed.
- H. O. Sintim, C. G. Mikek, M. Wang and M. A. Sooreshjani, MedChemComm, 2019, 10, 1999–2023 RSC.
- C. W. Karanja, K. S. Yeboah, W. W. S. Ong and H. O. Sintim, RSC Chem. Biol., 2021, 2, 206–214 RSC.
- C. Shu, G. Yi, T. Watts, C. C. Kao and P. Li, Nat. Struct. Mol. Biol., 2012, 19, 722–725 CrossRef CAS PubMed.
- N. Dayal, E. Řezníčková, D. E. Hernandez, M. Peřina, S. Torregrosa-Allen, B. D. Elzey, J. Škerlová, H. Ajani, S. Djukic, V. Vojáčková, M. Lepšík, P. Řezáčová, V. Kryštof, R. Jorda and H. O. Sintim, J. Med. Chem., 2021, 64, 1098–10996 CrossRef PubMed.
- K. E. Sivick, A. L. Desbien, L. H. Glickman, G. L. Reiner, L. Corrales, N. H. Surh, T. E. Hudson, U. T. Vu, B. J. Francica, T. Banda, G. E. Katibah, D. B. Kanne, J. J. Leong, K. Metchette, J. R. Bruml, C. O. Ndubaku, J. M. McKenna, Y. Feng, L. Zheng, S. L. Bender,C., Y. Cho, M. L. Leong, A. Elsas, T. W. Dubensky Jr. and S. M. McWhirter, Cell Rep., 2018, 25, 3074–3085 CrossRef CAS PubMed.
- S. K. Yebaoh, A. Zigli. and H. O. Sintim, ChemBioChem, 2024, e202400321 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03228d |
| This journal is © The Royal Society of Chemistry 2024 |