
Self-trapped emissions in 2D lead-free halide perovskites driven by divalent spacer cations†
Andre L. M.
Freitas
 *a,
Aryane
Tofanello
a,
Naidel A. M. S.
Caturello
b,
Karla K. F.
Barbosa
a and
Fabio F.
Ferreira
a
*a,
Aryane
Tofanello
a,
Naidel A. M. S.
Caturello
b,
Karla K. F.
Barbosa
a and
Fabio F.
Ferreira
a
aUniversidade Federal do ABC (UFABC), CCNH, Santo André-SP, Brazil. E-mail: andre_luizmf@yahoo.com.br; freitas.andre@ufabc.edu.br
bThomas Lord Department of Mechanical Engineering and Materials Science, Duke University, Durham, North Carolina 27708, USA
First published on 30th July 2024
Abstract
The mono- and divalent spacer cation investigation in sodium/indium-based 2D double perovskites revealed significant impacts on optoelectronic properties due to distortions in the inorganic layers. The strong electron–phonon coupling in a novel lead-free Dion–Jacobson phase highlights a promising class for broad-emission devices based on self-trapped excitons (STE), offering enhanced structural stability.
Rapid advancements in halide perovskites are crucial for optoelectronic devices, but challenges such as ion migration, structural instability, and toxicity still persist.1 Although 3D Pb-based halide perovskites show high electronic density and symmetry, their application in photovoltaics is hindered by degradation, high ionic conductivity,2 and moisture sensitivity.3 Consequently, extensive research has triggered the scientific community to find alternatives to these 3D structures. A well-studied approach involves utilizing 2D layered structures with organic spacer cations to confine inorganic entities. The best studied 2D halide perovskites are: (i) 2D Ruddlesden–Popper (2D-RP), which feature double layers of protonated organic molecules (R–NH3+) forming van der Waals gaps,4 and (ii) 2D Dion–Jacobson (2D-DJ), which have a single layer of bi-protonated organic molecules (NH3+–R–NH3+) that eliminate the van der Waals gap, promoting direct connectivity between inorganic layers and exhibiting greater structural robustness.5 Low-dimensional halide perovskites are notable for their tunable bandgaps,6 structural stability,7 and strong electron–phonon coupling,8 resulting in pronounced emission properties.9 These features made them promising candidates for optoelectronic devices compared to their 3D counterparts. The hydrophobic organic spacer notably prevents the degradation of inorganic sheets, enhancing environmental stability.
Aligned with the structural confinement in 2D perovskites, the degree of rotation and tilting of [BX6]4− inorganic octahedrons is responsible for tuning the optoelectronic properties in these materials. Furthermore, the chemical nature of the organic spacer and the interaction with the halide component are also decisive for the core properties of these materials. Undoubtedly, the presence of lead in the main reports in the area is still under debate due to its high toxicity. The substitution of Pb with nontoxic divalent metals that maintain excellent semiconducting properties is a topic of ongoing discussion; however, their susceptibility to oxidation affects perovskite efficiency and stability, which demand a careful analysis of the benefits of these alternatives.10,11 Under these conditions, double perovskites have shown robustness, achieving efficiencies comparable to Pb-based materials. Herein, this study aims to merge the benefits of layered perovskites with lead-free alternatives. We present advancements in synthesizing lead-free 2D double perovskites and explore different spacers to understand how cationic motifs influence their physicochemical properties. An attempt was made to describe the role of spacer cation configuration on octahedral distortions and excitonic properties using both experimental and theoretical approaches.
Specifically, a monovalent cation (Na+) and a trivalent cation (In3+) were used to achieve the charge balance necessary for the perovskite thermodynamic stability. The synthesis procedure was adapted from a previously reported method for synthesizing Na–In-based perovskite with stoichiometry (PEA)4NaInCl8.12,13 In our approach, the samples were prepared using a precipitation method at a temperature of 110 °C under stirring, followed by the rapid incorporation of organic molecules (details in the ESI†). Two different molecules were employed: (I) phenylethylamine (PEA), a mono-protonated amine with an aromatic ring, to form 2D-RP structures characterized by large interlayer space; and (II) 1,3-diaminopropane (DAP), a bi-protonated single-chain amine, aimed to form 2D-DJ structures with shorter interlayer and enhanced interaction between adjacent inorganic layers representing a novel approach in lead-free 2D perovskites.
By performing powder X-ray diffraction (PXRD), we confirmed the phase and structure of the synthesized samples, both with low dimensionality, n = 1 (Fig. 1). Under ambient conditions, 2D-RP sample, (PEA)4NaInCl8, fits in the monoclinic phase with the centrosymmetric space group P21/c (no 14) with lattice parameters a = 7.3924(6) Å, b = 39.42(4) Å, c = 7.3492(6) Å, and β = 90.326°, displayed in the Fig. 1a, in accordance with the previously reported in the literature.12 PXRD analysis for (PEA)4NaInCl8 was performed on a macerated sample, and to observe orientation differences, we present the refinement for the as-grown sample in Fig. S1 (ESI†). Similarly, for the novel 2D-DJ stoichiometry, (DAP)2NaInCl8, the obtained PXRD pattern (Fig. 1b) shows its characteristic diffraction peaks at 10.60, 11.05, 11.52, 13.13, 15.60,18.16, 18.51, 19.19 and 19.85° (2θ, λ = 1.54056 Å) corresponding to the (002), (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), (101), (011), (101), (110), (10
), (101), (011), (101), (110), (10![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ), and (11
), and (11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) planes, respectively. This indicates that (DAP)2NaInCl8 consists of a monoclinic phase, indexed to the P21 space group with lattice parameters a = 8.8531(6) Å, b = 7.3328(9) Å, c = 16.748(7) Å, and β = 93.46(4)°. The phase was relaxed by theoretical calculations, which showed consistent agreement with experimental data. The schematic structure shown in Fig. 1c illustrates that the combined crystallographic arrangement and the short covalent interlayer spacing in the DJ structure led to a reduced distance between adjacent octahedral layers.
) planes, respectively. This indicates that (DAP)2NaInCl8 consists of a monoclinic phase, indexed to the P21 space group with lattice parameters a = 8.8531(6) Å, b = 7.3328(9) Å, c = 16.748(7) Å, and β = 93.46(4)°. The phase was relaxed by theoretical calculations, which showed consistent agreement with experimental data. The schematic structure shown in Fig. 1c illustrates that the combined crystallographic arrangement and the short covalent interlayer spacing in the DJ structure led to a reduced distance between adjacent octahedral layers.
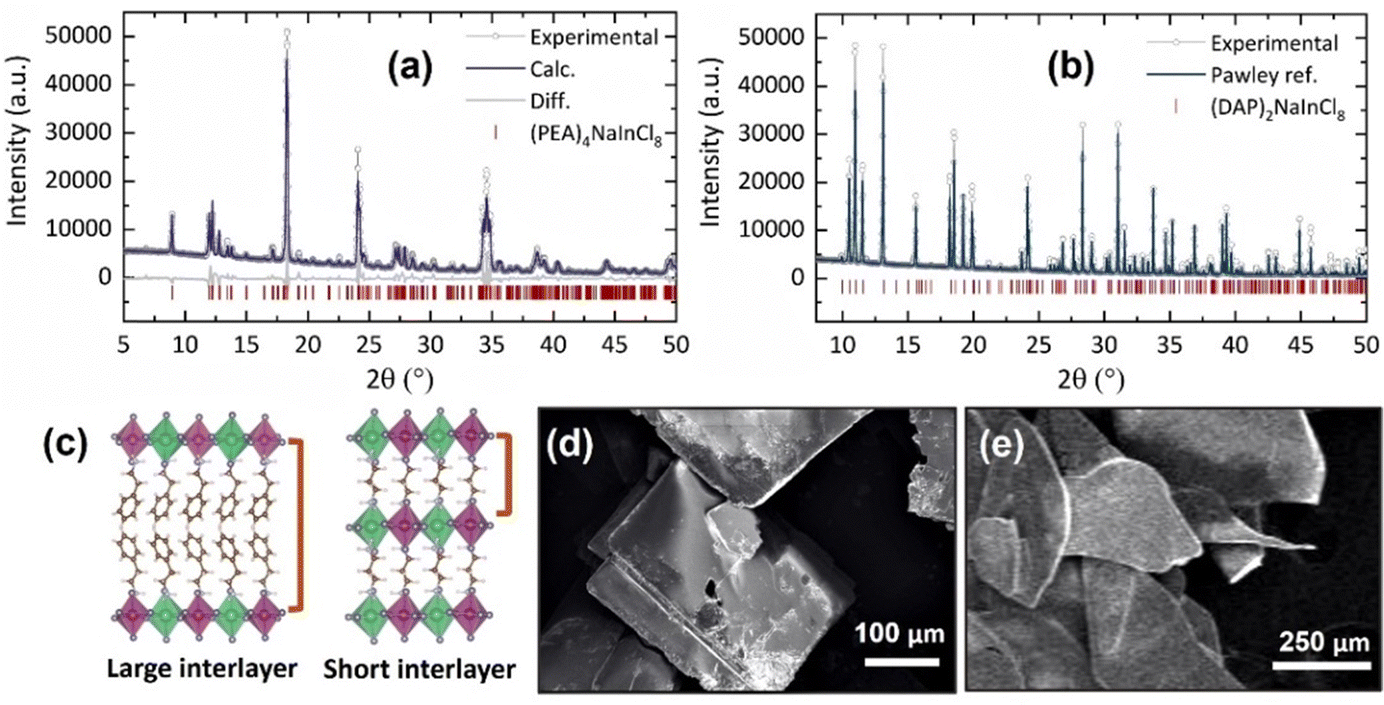 | ||
| Fig. 1 Powder X-ray diffraction patterns (PXRD): (a) Rietveld refinement of (PEA)4NaInCl8; (b) Pawley refinement of (DAP)2NaInCl8. (c) Schematic representation of the crystal structure at the organic–inorganic interface showing large and small interlayer spacings. Scanning electron microscopy (SEM) images of (d) (PEA)4NaInCl8 and (e) (DAP)2NaInCl8. | ||
SEM images confirmed the lamellar morphologies of both samples. The 2D-RP sample exhibited well-defined square plates (Fig. 1d). Similarly, (DAP)2NaInCl8 showed a lamellar morphology with less defined growth in the xy-plane (Fig. 1e) and colorless thin crystals (Fig. S2, ESI†). This suggests that the incorporation of the DAP and the resulting change in the chemical environment during synthesis modify the long-range symmetry in the crystal growth mechanism, preventing the formation of well-defined square plates. Additionally, density-functional theory (DFT) calculations of angle distortions in the axial and equatorial planes reveal considerable local asymmetries in the (DAP)2NaInCl8 sample (Table 1 and Fig. 2a). This suggests that the bi-protonated molecule promotes greater distortions by directly interacting with two adjacent inorganic layers. Because the system retains a global centrosymmetric space group, it does not exhibit Rashba-type spin splitting at any path of the Brillouin zone for conduction-band- or valence-band-derived bands, Fig. S3 (ESI†). Fig. S4a (ESI†) shows that the conduction band mainly comes from the anti-bonding halogen np and In 5p orbitals. In contrast, the valence band primarily derives from the halogen p orbitals along the axial direction. The orbital composition is evident from the partial density of states (pDOS) shown in Fig. S4b (ESI†). The presence of anti-bonding orbitals in the edge bands leads to increased repulsion due to local asymmetries, which is the main mechanism for the emergence of a large [HSE06 + SOC] band gap of 4.95 eV for (DAP)2NaInCl8, thus leading the synthesized DJ phase to be expected to be a wide-bandgap semiconductor.
| System | (DAP)2NaInCl8 | (PEA)4NaInCl8 |
|---|---|---|
| Δβin (°) | 8.53 | 0.45 |
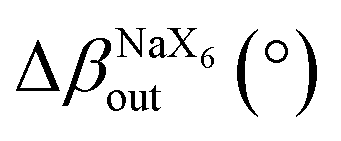
|
12.87 | 0.56 |
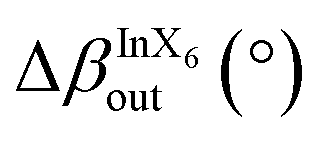
|
12.14 | 0.56 |
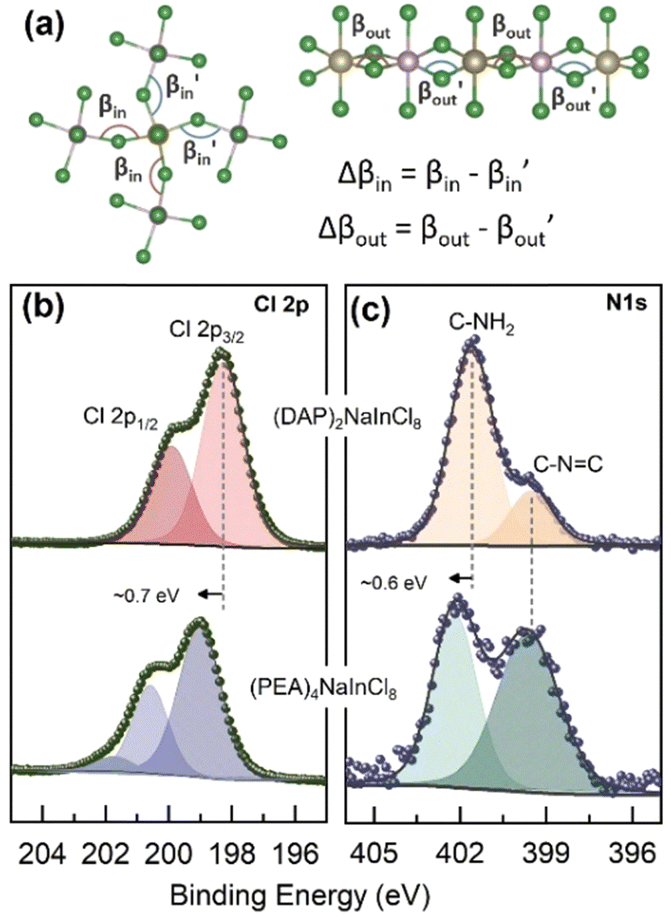 | ||
| Fig. 2 (a) Schematic showing the asymmetric in-plane and out-of-plane tilting within the inorganic parts of the perovskites along with their mathematical definitions used to build Table 1. XPS core-level spectra of (b) Cl 2p and (c) N 1s of the 2D perovskite (DAP)2NaInCl8 (upper panel) and (PEA)4NaInCl8 (lower panel). | ||
X-ray photoelectron spectroscopy (XPS) was employed to gain insights into the surrounding chemical environment at the interface between the organic spacer and the halide within the inorganic octahedron, utilizing a monochromatic Al Kα X-ray source. The high-resolution spectra for Cl 2p and N 1s are displayed in Fig. 2b and Fig. 2c, respectively. The Cl 2p core-level spectrum (Fig. 2b) revealed a binding energy shift of approximately 0.7 eV between (DAP)2NaInCl8 (upper panel) and (PEA)4NaInCl8 (lower panel). Such energy shift suggests a change in the chlorine atoms neighbors.
As expected for 2D structures, the penetration of the organic molecule into the octahedral plane and the formation of hydrogen bonds can significantly modify the chemical environment. High-resolution N 1s spectrum in Fig. 2c shows a peak at ∼399.6 eV in both perovskites, attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH2+ interactions.14 However, a higher energy peak was also observed (∼401.6 eV), corresponding to the amino group (NH2) in the organic molecules. This peak exhibited a shift of approximately 0.6 eV (∼402.2 eV) between the different structures. Studies using NMR indicate that the rotational rigidity of NH3 groups correlates with the chemical nature of the spacers and is directly associated with octahedral inclination.15 The balance of Coulombic and hydrogen-bond interactions with octahedra and van der Waals forces (in the RP structure) significantly influences the local dynamics. Consequently, the rotational rigidity of the covalently locked spacer in the DJ structure directly affects interlayer interactions.16 Similarly, Li et al.17 experimentally observed that spacers and interface interactions notably alter the electronic density around the B (or B′) site and the halide due to the strong conjugation of these molecules. Notably, the lower binding energy values observed for the (DAP)2NaInCl8 suggest a higher electron density. Thus, the XPS shifts observed here, among different structures, can be linked to changes in the interactions within the local chemical environment.
NH2+ interactions.14 However, a higher energy peak was also observed (∼401.6 eV), corresponding to the amino group (NH2) in the organic molecules. This peak exhibited a shift of approximately 0.6 eV (∼402.2 eV) between the different structures. Studies using NMR indicate that the rotational rigidity of NH3 groups correlates with the chemical nature of the spacers and is directly associated with octahedral inclination.15 The balance of Coulombic and hydrogen-bond interactions with octahedra and van der Waals forces (in the RP structure) significantly influences the local dynamics. Consequently, the rotational rigidity of the covalently locked spacer in the DJ structure directly affects interlayer interactions.16 Similarly, Li et al.17 experimentally observed that spacers and interface interactions notably alter the electronic density around the B (or B′) site and the halide due to the strong conjugation of these molecules. Notably, the lower binding energy values observed for the (DAP)2NaInCl8 suggest a higher electron density. Thus, the XPS shifts observed here, among different structures, can be linked to changes in the interactions within the local chemical environment.
Furthermore, previous studies18 have shown that interstitial dopants can cause lattice deformations and larger octahedral tilting, leading to shifts in XPS binding energy. Liang et al.19 showed that lattice strain accumulates within the inorganic layers but is dampened by the organic layers. In low-dimensional structures, the strain extends both in-plane and out-of-plane. The organic layer allows rotational freedom for the octahedra, leading to the greatest distortions observed in these n = 1 perovskites. The (DAP)2NaInCl8 structure, with a single spacer layer connecting two adjacent octahedral layers, promotes a higher degree of distortion in the inorganic content. Compared to the PEA molecule, which has significant rotational freedom around its ethylammonium tail, the DAP cation exhibits restricted rotation and increased steric bulk within the interlayer. This distortion, confirmed by structural analysis and theoretical calculations, plays a crucial role in shaping the optoelectronic properties.
In this context, the optical characterization was performed using photoluminescence (PL) spectroscopy, as shown in Fig. 3a. With the structural change, the static PL spectra emission peak shifts by about 103 nm. (PEA)4NaInCl8 exhibits an asymmetric peak with strong intensity around 283 nm, while (DAP)2NaInCl8 emits at ∼386 nm. Further, we investigated the excitation spectra (PLE), where the 2D-RP phase (Fig. 3b) shows an emission independent of the excitation. Such behavior indicates that the exciton recombination dynamics stem from a single excited state, primarily band-to-band transition. Conversely, the 2D-DJ phase (Fig. 3c) exhibits an excitation-dependent emission shift, indicating that the excitation–emission processes originate from different excited states.20–22 In the schematic Fig. 3d and e, it is shown that the emission mechanism differs between (PEA)4NaInCl8 and (DAP)2NaInCl8 due to sub-gap states, with the latter exhibiting emission from sub-gap states. The intrinsic lattice distortion in the 2D-DJ sample (Fig. 3e) promotes recombination dominated by self-trapped exciton states (STE), enabling the tuning of optical properties based on excitation. These excitation-dependent transitions result in a broadband emission mechanism. In other words, the intrinsic distortions in the DJ structure trap free excitons in states with an energy barrier below the gap due to increased exciton–phonon coupling in the inorganic layer.23,24 Thus, the emission process is dominated by STE states, resulting in radiative recombination with energies lower than the bandgap energy. Specifically, our results indicate that the photoexcitation and emission processes in these low-dimensional perovskites are significantly influenced by the organic–inorganic interface and its resulting distortions and structural configurations. These factors directly affect the optoelectronic structure–property relationships, aligning with discussions in previous reports.25,26
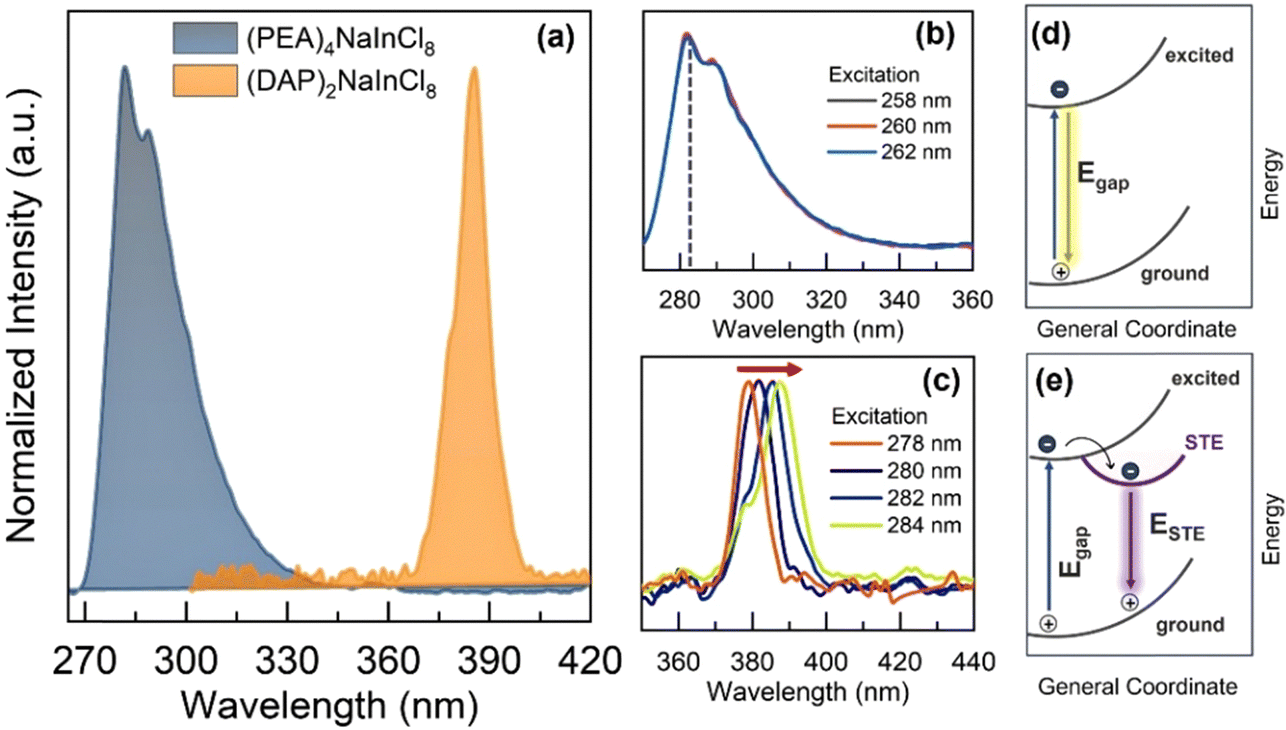 | ||
| Fig. 3 (a) Normalized PL-intensity of two-dimensional double perovskites; excitation-dependent photoluminescence spectra (PLE) for: (PEA)4NaInCl8 and (DAP)2NaInCl8, (b) and (c) respectively. Schematic sketch of radiative recombination in the 2D-RP structure (d) and 2D-DJ structure (e). | ||
The dynamics of organic spacers can significantly modify the local chemical environment, including local distortions and potential differences, leading to changes in physical properties. Additionally, the softening effect caused by the spacers enhance the electron–phonon coupling in 2D perovskites, strongly impacting carrier dynamics.27 In summary, we have developed a new lead-free double-layered perovskite material with excellent stability and optical properties under ambient conditions. By reducing dimensionality and introducing intrinsic distortions in the inorganic lattice, we achieve emissions dominated by self-trapped states, leading to adjustable optical properties. Lead-free halide perovskite has gained attention for its low toxicity and wide range of applications, including LEDs, detectors, and scintillators, covering nearly all areas of optoelectronics.28 Thus, our study provides new perspectives into underexplored 2D double perovskites with potential for solid-state optoelectronic applications.
This work received financial support from FAPESP (grants 2022/06433-5 and 2023/12165-6). The authors would like to acknowledge the experimental support from the Multiuser Central Facilities (UFABC). A. T. acknowledges the additional support provided by the Human Resource Program of The Brazilian National Agency for Petroleum, Natural Gas, and Biofuels – PRH-ANP 49 (grant 045919) and CNPq/MCTI 406470/2022-7. N. A. M. S. C. acknowledges Molecular Simulations from First Principles (MS1P) e. V. for his fellowship.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- S. Wu, Z. Chen, H. L. Yip and A. K. Y. Jen, Matter, 2021, 4, 3814–3834 CrossRef CAS.
- A. L. M. Freitas, A. Tofanello, A. Bonadio and J. A. Souza, J. Mater. Sci.: Mater. Electron., 2022, 33, 18327–18344 CrossRef CAS.
- B. P. Kore, M. Jamshidi and J. M. Gardner, Mater. Adv., 2024, 5, 2200–2217 RSC.
- C. C. Stoumpos, D. H. Cao, D. J. Clark, J. Young, J. M. Rondinelli, J. I. Jang, J. T. Hupp and M. G. Kanatzidis, Chem. Mater., 2016, 28, 2852–2867 CrossRef CAS.
- X. Li, W. Ke, B. Traoré, P. Guo, I. Hadar, M. Kepenekian, J. Even, C. Katan, C. C. Stoumpos, R. D. Schaller and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 12880–12890 CrossRef CAS PubMed.
- F. Nayab, M. Aamir, M. E. Khan, Q. Wali, M. Sher, H. Khurshid and J. Akhtar, Phys. Chem. Chem. Phys., 2024, 26, 6058–6067 RSC.
- K. Meng, X. Wang, Z. Li, Z. Liu, Z. Qiao, C. Wang, Y. Hu, S. Li, L. Cheng, Y. Zhai and G. Chen, Energy Environ. Sci., 2021, 14, 2357–2368 RSC.
- K. M. McCall, C. C. Stoumpos, S. S. Kostina, M. G. Kanatzidis and B. W. Wessels, Chem. Mater., 2017, 29, 4129–4145 CrossRef CAS.
- L. Zhang, Y. Liu, Z. Yang and S. (Frank) Liu, J. Energy Chem., 2019, 37, 97–110 CrossRef.
- J. Wang, J. Dong, F. Lu, C. Sun, Q. Zhang and N. Wang, J. Mater. Chem. A, 2019, 7, 23563–23576 RSC.
- W. Ke, C. C. Stoumpos, M. G. Kanatzidis, W. Ke, C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2019, 31, 1803230 CrossRef CAS PubMed.
- Y. Zhang, X. Liu, H. Sun, J. Zhang, X. Gao, C. Yang, Q. Li, H. Jiang, J. Wang and D. Xu, Angew. Chem., Int. Ed., 2021, 60, 7587–7592 CrossRef CAS PubMed.
- J. Xue, Z. Wang, A. Comstock, Z. Wang, H. H. Y. Sung, I. D. Williams, D. Sun, J. Liu and H. Lu, Chem. Mater., 2022, 34, 2813–2823 CrossRef CAS.
- Q. Zhou, L. Liang, J. Hu, B. Cao, L. Yang, T. Wu, X. Li, B. Zhang and P. Gao, Adv. Energy Mater., 2019, 9, 1802595 CrossRef.
- C. J. Dahlman, R. M. Kennard, P. Paluch, N. R. Venkatesan, M. L. Chabinyc and G. N. Manjunatha Reddy, Chem. Mater., 2021, 33, 642–656 CrossRef CAS.
- C. T. Triggs, R. D. Ross, W. Mihalyi-Koch, C. F. M. Clewett, K. M. Sanders, I. A. Guzei and S. Jin, ACS Energy Lett., 2024, 9, 1835–1843 CrossRef CAS.
- B. Li, Q. Liu, J. Gong, S. Li, C. Zhang, D. Gao, Z. Chen, Z. Li, X. Wu, D. Zhao, Z. Yu, X. Li, Y. Wang, H. Lu, X. C. Zeng and Z. Zhu, Nat. Commun., 2024, 15, 1–9 Search PubMed.
- P. Yadav, D. Gill, S. Khurana, R. S. Lamba, S. Bhattacharya and S. Sapra, J. Phys. Chem. C, 2022, 127, 34 Search PubMed.
- M. Liang, W. Lin, Q. Zhao, J. Li, L. Zhu, B. Sarpi, A. Zakharov, I. G. Scheblykin, T. Pullerits, Y. Niu, S. E. Canton and K. Zheng, Sol. RRL, 2023, 7, 2200795 CrossRef CAS.
- S. Kahmann, D. Meggiolaro, L. Gregori, E. K. Tekelenburg, M. Pitaro, S. D. Stranks, F. De Angelis and M. A. Loi, ACS Energy Lett., 2022, 7, 4232–4241 CrossRef CAS PubMed.
- J. Tan, D. Li, J. Zhu, N. Han, Y. Gong and Y. Zhang, Nanoscale, 2022, 14, 16394–16414 RSC.
- J. Li, H. Wang and D. Li, Front. Optoelectron., 2020, 13, 225–234 CrossRef PubMed.
- M. Seitz, A. J. Magdaleno, N. Alcázar-Cano, M. Meléndez, T. J. Lubbers, S. W. Walraven, S. Pakdel, E. Prada, R. Delgado-Buscalioni and F. Prins, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- M. D. Smith, B. A. Connor and H. I. Karunadasa, Chem. Rev., 2019, 119, 3104–3139 CrossRef CAS PubMed.
- A. Tofanello, A. L. M. Freitas, T. B. de Queiroz, A. Bonadio, H. Martinho and J. A. Souza, J. Phys. Chem. Lett., 2022, 13, 1406–1415 CrossRef CAS PubMed.
- A. L. M. Freitas and J. A. Souza, J. Mater. Chem. C, 2023, 11, 6651–6661 RSC.
- J. Duan, J. Li, G. Divitini, D. Cortecchia, F. Yuan, J. You, S. Liu, A. Petrozza, Z. Wu, J. Xi, J. Duan, F. Yuan, Z. Wu, J. Xi and J. Li, Adv. Mater., 2024, 2403455 CrossRef CAS PubMed.
- S. A. Khan, N. Z. Khan, M. Sohail, M. Runowski, X. Xu and S. Agathopoulos, Mater. Today Phys., 2023, 34, 101079 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03192j |
| This journal is © The Royal Society of Chemistry 2024 |