
Photocatalytic decarboxylation of free carboxylic acids and their functionalization
Subal
Mondal†
a,
Subham
Mandal†
a,
Soumya
Mondal†
a,
Siba P.
Midya
b and
Pradyut
Ghosh
 *a
*a
aSchool of Chemical Sciences, Indian Association for the Cultivation of Science, Kolkata 700032, India. E-mail: icpg@iacs.res.in
bDepartment of Chemistry, Jadavpur University, 188 Raja S. C. Mullick Road, Kolkata 700032, India
First published on 1st August 2024
Abstract
Visible light mediated decarboxylative functionalization of carboxylic acids and their derivatives has recently emerged as a novel and powerful toolkit for small molecule activation in diverse carbon–carbon and carbon–hetero bond forming reactions. Naturally abundant highly functionalized bench-stable carboxylic acid analogs have been employed as promising alternatives to non-trivial organometallic reagents for mild and eco-benign synthetic transformation with traceless CO2 by-products. In this highlight article, we focus on the development of various photodecarboxylative functionalization strategies along with intra/inter-molecular cyclization via concerted single electron transfer (SET) or energy transfer (ET) pathways. Moreover, widely explored carboxylic acids are systematically classified here into four categories; i.e., α-keto, aliphatic, α,β-unsaturated, and aromatic analogs for a concise overview to the readership. The association of decarboxylative radical species with coupling partners to construct C–C and C–N/O/S/P/X bonds for each analogous acid has been presented in brief.
 Subal Mondal | Subal Mondal received his BSc degree in 2017 from Syamsundar College under the University of Burdwan in India. After completing his Master's degree in 2019 from IIT Guwahati, he joined PhD under the supervision of Prof. Pradyut Ghosh in IACS Kolkata, India. Currently, he is working as a senior research fellow at IACS Kolkata. His PhD research work is based on the development of new organic methodologies using photoredox catalysis. |
 Subham Mandal | Subham Mandal received his BSc degree in 2017 from Krishnath College under the University of Kalyani in India. He completed his Master's degree in 2019 from the University of Kalyani. He then joined PhD under the supervision of Prof. Pradyut Ghosh in IACS Kolkata, India. Currently, he is working as a senior research fellow at IACS Kolkata. His PhD research work is based on the development of a new macrocycle and macrobicycle for selective supramolecular catalysis. |
 Soumya Mondal | Soumya Mondal received his BSc degree in 2019 from Abhedananda Mahavidyalaya under the University of Burdwan in India. He joined IACS Kolkata for the Integrated MS/PhD programme in 2019. He has joined PhD under the supervision of Prof. Pradyut Ghosh in IACS Kolkata, India. Currently, he is working as a senior research fellow at IACS Kolkata. His PhD research work is based on the development of new cross-coupling reactions using photoredox catalysis. |
 Siba P. Midya | Dr Siba P. Midya completed his BSc from Bajkul Milani Mahavidyalaya under Vidyasagar University, India. He obtained his Master's degree from IIT Bombay (2013) and joined CSIR-NCL Pune for PhD in 2014 under Prof. E. Balaraman. After PhD degree in 2019, he joined as a Research Associate followed by a National Post-Doctoral Fellow (NPDF) under the guidance of Prof. Pradyut Ghosh at IACS Kolkata, India. Currently, he is a DST-INSPIRE Faculty at Jadavpur University, Kolkata. His research focuses on new methodologies using Earth-abundant metal based pincers and photoredox catalysts. |
 Pradyut Ghosh | Prof. Pradyut Ghosh is a senior professor in the School of Chemical Sciences at the Indian Association for the Cultivation of Science (IACS), India. He received his PhD in Chemistry from the Indian Institute of Technology, Kanpur under the direction of Parimal K. Bharadwaj in 1998. He spent two years as a post-doctoral fellow at Texas A&M University, with Richard M. Crooks and he was an Alexander von Humboldt Fellow at the University of Bonn, Germany in Fritz Vögtle's and Christoph A. Schalley group. Upon his return to India, he joined Central Salt & Marine Chemicals Research Institute, India and in 2007 he moved to IACS. His present research interests cover the designing of Interlocked Molecular Systems, Recognition, Sensing and Extraction of ions and ion-pairs, and Supramolecular and Photo-Catalysis. |
1. Introduction
Carboxylic acids are ubiquitous, structurally diverse and synthetically applicable as a synthon for important alkyl or aryl motifs in organic transformations.1–3 The release of CO2, known as decarboxylation, is a fundamental step in biological and chemical conversions for diverse carbon–carbon (C–C) and carbon–hetero (C–Het) bond formation.4 Enzyme catalysed bio-decarboxylation facilitates many important bio-transformations, such as the conversion of pyruvic acid into acetyl-CoA by the pyruvate dehydrogenase complex, which is a crucial step of the Krebs cycle.5,6 Inspired by Nature's toolbox, synthetic chemists have utilised decarboxylation protocols toward the regio-selective generation of highly reactive radical species to access complex molecularity.7,8 Kolbe electrolysis9 and Cu/Ag salt mediated thermal decarboxylations10,11 are the earliest methods for CO2 extrusion with limited applicability due to harsh conditions and low regio-selectivity. The subsequent advancement of transition metal-mediated decarboxylation was reported by the Sheppard, Nilsson and Myers groups from the mid-1900s but those suffered from the usage of stoichiometric metal salts and limited scope.12–14 In 2006, Goossen and co-workers reported the first example of catalytic decarboxylation to access bi-aryl scaffolds using a bimetallic Pd/Cu system.15 Thereafter, a significant advancement in transition metal catalysed decarboxylation for diverse C–C and C–hetero bond formation has gained much attention over the past decades.16–19 However, the high temperature conditions and stoichiometric oxidant usage demand a green and sustainable catalytic pathway towards decarboxylative bond forming reactions.Most recently, photocatalytic single electron or energy transfer has emerged as one of the fundamental synthetic protocols for remote functionalization, regio-selective cross-coupling reactions and C–H bond activations.20–23 Subsequently, the merging of photoredox with transition metals, known as metallaphotoredox catalysis, unveiled a new mechanistic pathway that reconfigures the synthetic library to access unusual and complex molecular scaffolds.8,24,25 Utilizing these photocatalytic plethoras, bench-stable aliphatic or aromatic carboxylic acids are functionalized by decarboxylation towards C–C/hetero bond formation at room temperature and under oxidant free conditions.26–28
To the best of our knowledge, the first photocatalytic decarboxylation was developed by Nishibayashi29 and Lei30 groups independently in 2013 using Ru and Ir photocatalysts. In the subsequent year, the MacMillan group reported decarboxylative arylation of α-amino acids via visible light mediated photoredox catalysis for C(sp2)–C(sp3) cross-coupling.31 A novel aliphatic carboxylic acid variant, known as acrylic acid, also underwent decarboxylation in the presence of hypervalent iodine towards a C(sp2)–C(sp3) cross-coupling reaction under blue light-emitting diode (LED) irradiation.32 These pioneering developments for aliphatic acid decarboxylation have leveraged new modes of coupling reactions for the construction of potent drugs and bio-relevant molecules. The remaining aryl carboxylic acid analogs are also bench-stable, structurally diverse precursors for various building blocks found in nature and commercial sources. Typically, the rate of decarboxylative aryl radical formation is lower than that of aliphatic carboxylic acids (kAro = 106 s−1vs. kAli = 109 s−1).33 Thus, aromatic acid decarboxylation is difficult to harness with the abovementioned photocatalytic methods. Most recently, the Ritter group has demonstrated a visible light-mediated ligand-to-metal-charge-transfer (LMCT) protocol to generate open-shell aryl radical species with low energy barrier and it opens up a new doorway of aromatic carboxylic acid functionalization.34
Overall, remarkable progress in the field of photocatalytic decarboxylation reactions has been devoted by the scientific community throughout the globe. To date, several reviews have been reported on the decarboxylative bond formations and few on substrate selective photo-decarboxylation reactions.26,27,31,35 However, the complete development of photocatalytic decarboxylation has not been summarised in a single review. In this article, we present the development of visible light mediated decarboxylation reactions that have elevated vast modes of C–C, C–O, C–N, C–P and C–S cross-couplings, cyclizations, and functionalization. Regardless of various important architectural motifs, we have tried to cover up all varieties of carboxylic acids by dividing the substrate analogs into four categories; i.e., α-keto acids; cyclic and acyclic aliphatic acids; α,β-unsaturated acids and aromatic acids. We are aware of numerous types of mechanistic pathways to construct a complex photocatalytic architecture, which is beyond the scope of this article. This highlight article intends to cover the year-wise evaluation of photo-decarboxylative functionalization, which will provide a comprehensive bond forming overview to the readers (Fig. 1).
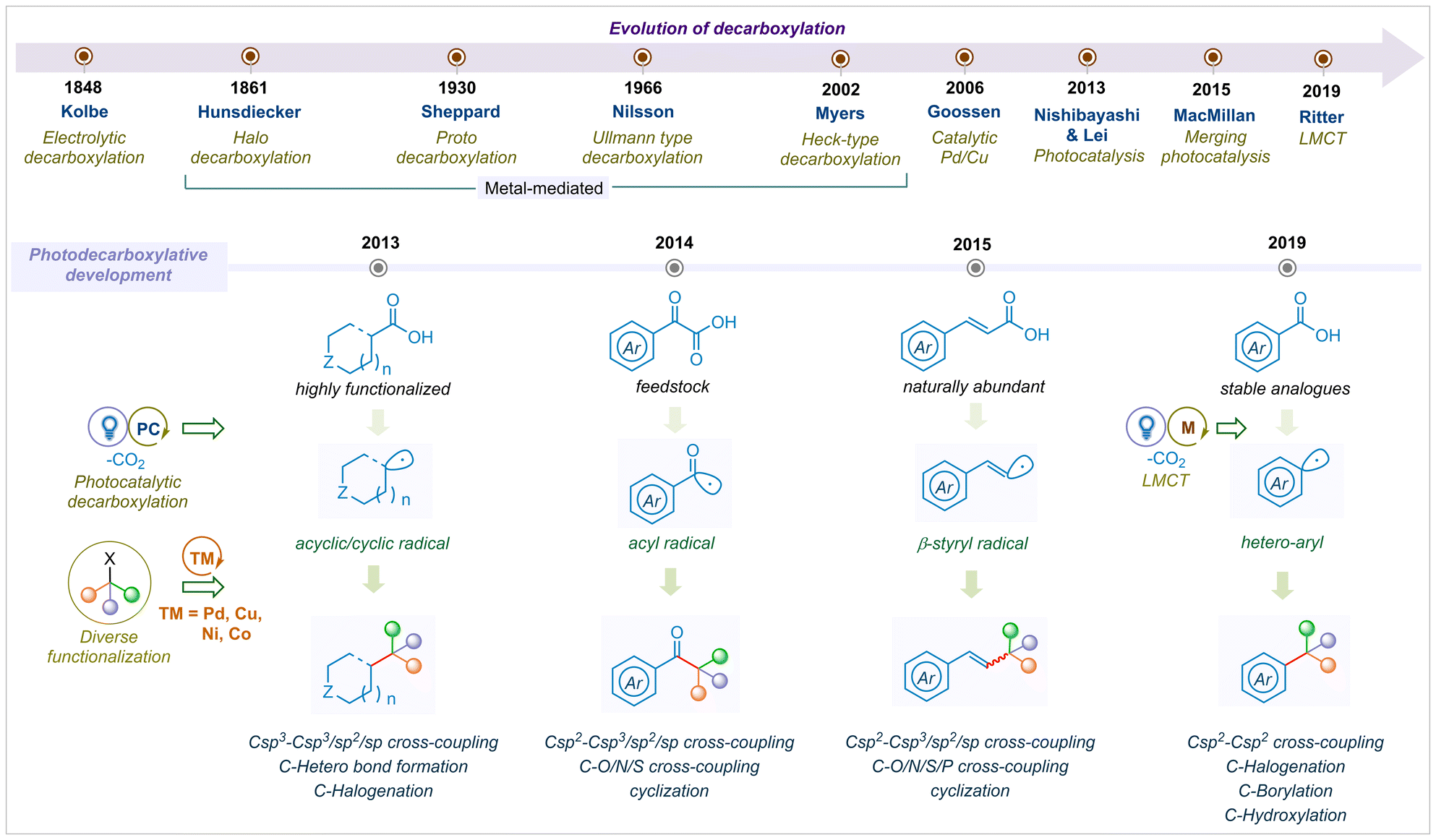 | ||
| Fig. 1 Evolution of decarboxylation from electrocatalysis to photocatalysis for diverse functionalization. | ||
2. Decarboxylation of α-keto acids
α-keto acids, a class of organic carboxylic acids containing the keto group at the α-position are significant acyl synthons used in synthetic organic chemistry. However, the decarboxylation of α-keto acids is limited to high temperatures and the usage of strong oxidants. In 2014, Lei and co-workers revealed an amazing breakthrough in the decarboxylation of carboxylic acids under photocatalytic conditions.302.1 C–C cross-coupling and cyclization
In 2008, Goossen and co-workers reported the first catalytic method for decarboxylative cross-coupling of α-keto acids with aryl halides using a Pd/Cu dual catalyst.36 Due to the requirement of a high temperature, this reaction is incompatible with the synthesis of thermally labile organic compounds. Following Lei and Lan's pioneering work on photoredox-catalyzed decarboxylative amidation, MacMillan and his team developed a mild and straightforward method for synthesizing ketones (2.1). This approach involved the decarboxylation of α-keto acids for cross-coupling with aryl bromides, utilizing a combined Ir/Ni photocatalytic system (Fig. 2a).37 Of late, several groups have explored versatile modes of arylation with α-keto acids and different aryl halides or boronic acids under different photocatalytic conditions.38–42 In 2015, Chen and co-workers demonstrated decarboxylative ynonylation using a [Ru(bpy)3](PF6)2 photocatalyst and hypervalent iodine(III) reagents (HIR).43 This reaction represents the first example of merging photoredox/HIR catalysis for decarboxylation of α-keto acids, which leads to the formation of acyl radicals and their unprecedented addition to HIR-bound alkynes (Fig. 2b).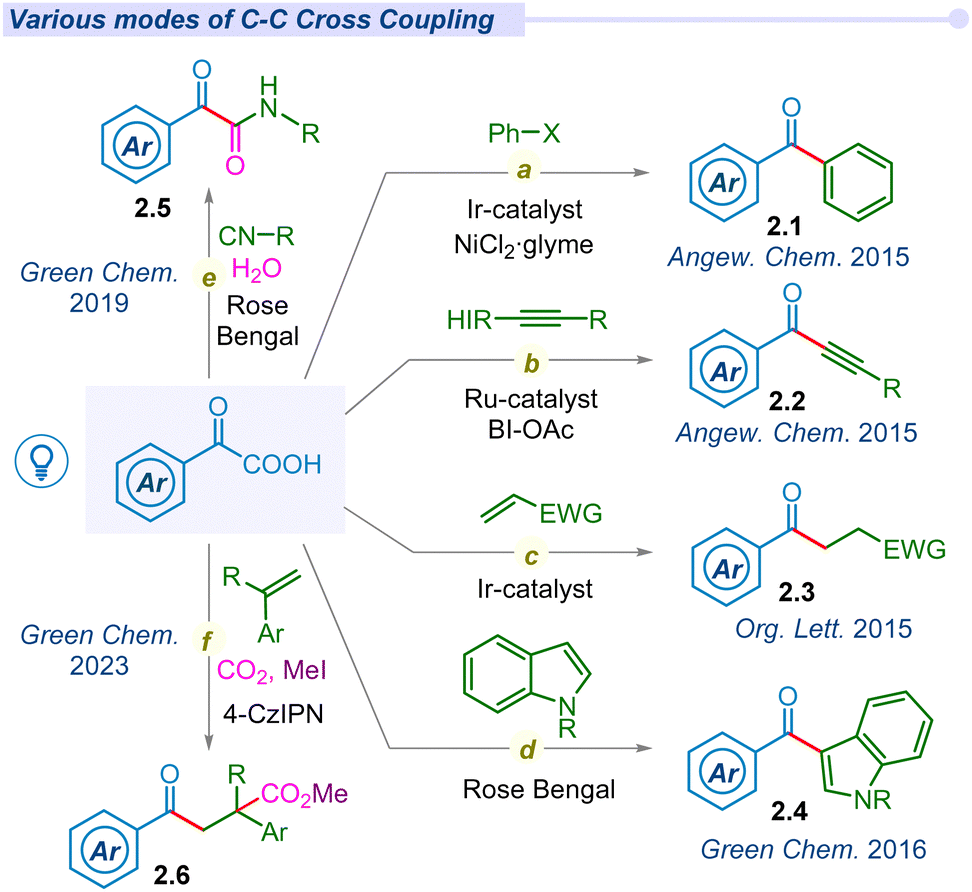 | ||
| Fig. 2 Decarboxylative C–C cross-coupling of α-keto acids. | ||
In 2015, Zu et al. explored the decarboxylative acylation of α-keto acids with electron-deficient olefins under visible-light induced photoredox catalysis (Fig. 2c).44 They successfully employed this acyl radical to a series of Michael acceptors including aldehydes, ketones, amides, nitriles, α,β-unsaturated esters and sulfones towards 1,4-conjugate addition. Later, similar modes of transformation have been developed by several groups.45–49 In the same year, Wang and co-workers proposed a novel approach for ortho acylation of acetanilide derivatives through decarboxylation of α-keto acids under a dual Eosin-Y/Pd-catalytic cycle.50 α-Keto acids have been employed for C(sp2)–C(sp3) cross-coupling through C–H acylation of azo and azoxybenzene moieties, using acridinium salt as an effective organophotocatalyst under blue LED irradiation.51
In 2016, Wand and Gu independently synthesized 3-acylindoles (2.4) from feedstock indoles and α-keto acids under photocatalytic conditions (Fig. 2d).52,53 In 2019, Wei and co-workers proposed a potent method for the synthesis of α-ketoamides (2.5) using α-keto acids, isocyanides and water as precursors under visible light conditions using the Rose Bengal photocatalyst (Fig. 2e).54 These mild and metal-free conditions offer a unique and straightforward method for preparing α-ketoamides through decarboxylation, radical addition and hydration processes in a cascade manner from feedstock chemicals. Thereafter, Prabhu and co-workers applied this protocol for the acylation of various electron-deficient heteroarenes, such as pyridines, quinolones, isoquinoline and phenanthridine, to their respective analogs under visible light irradiation.55 Similarly, the He group established decarboxylative acylation of quinoxalin-2-(1H)-ones using aerial oxygen as a sole oxidant under visible light conditions.56 However, this photo-enabled protocol did not require any external photosensitizers, powerful oxidants or metal catalysts. The Chu group has developed Minisci-type reactions for pyridine N-oxides via decarboxylative cross-coupling using a new generation organophotocatalyst fluorescein dimethyl ammonium.57
Jana and co-workers developed a novel strategy for regio-selective di-functionalization at the α- and β-positions of styrenes using CO2 under metal-free conditions (Fig. 2f).58 Mechanistic investigation revealed radical–radical cross-coupling and radical polar cross-over for acylative-benzylation and acylative-carboxylation to produce benzylic carbanions, which further act as nucleophiles towards carbon dioxide molecules.
Substantial development in the decarboxylative acylation of α-keto acids has leveraged the synthetic route of cross-couplings over the years. Apart from these seminal works, several groups have designed various efficient routes to employ the ‘acyl’ radical in one pot intra/inter-molecular cyclization reactions (Fig. 3). In 2016, Wang and co-workers unveiled a catalyst-free visible light irradiated approach for carbonyl-arylation of acrylamides with α-keto acids using HIR at room temperature (Fig. 3a).59 Similarly, Chu et al. reported a one-pot photo-induced decarboxylative reaction for the synthesis of 4-aryl-2-quinolinones (3.2) through an intramolecular cyclization (Fig. 3b).60 In 2020, the Yao group utilized the energy of visible light to develop an efficient route to synthesize 2-acylindoles (3.3) through decarboxylative cyclization of 2-alkenylarylisocyanides (Fig. 3c).61 Mechanistic insights unfolded that the reaction proceeded through the sequential addition of acyl radical to 2-alkenylarylisocyanides, followed by the 5-exo-trig cyclization. The Miao group developed a photosensitizer-free decarboxylative cyclization reaction of 2-isocyanobiphenyls to access phenanthridin-6-yl(aryl)methanol (3.4) in a cascade manner (Fig. 3d).62 Mechanistic analysis found that the decarboxylation process occurred via the formation of an electron donor–acceptor complex (EDA) under visible light irradiation. Su et al. utilized and employed Eosin-B and (NH4)2S2O8 oxidant for synthesizing acylated isoquinoline-1,3(2H,4H)-dione (3.5) derivatives through visible-light mediated decarboxylative cyclization of N-methacryloyl-N-methylbenzamide and α-keto acids (Fig. 3e).63 In 2021, the Yu group disclosed a metal-free decarboxylative cyclization method for the construction of acylated heterocyclic derivatives (3.6).64 They successfully synthesized indolo[2,1-a]isoquinolin-6(5H)-one, benzimidazo, thioflavone, aroylazaspiro[4.5]trienone derivatives using 4-CzIPN as a photocatalyst (Fig. 3f).
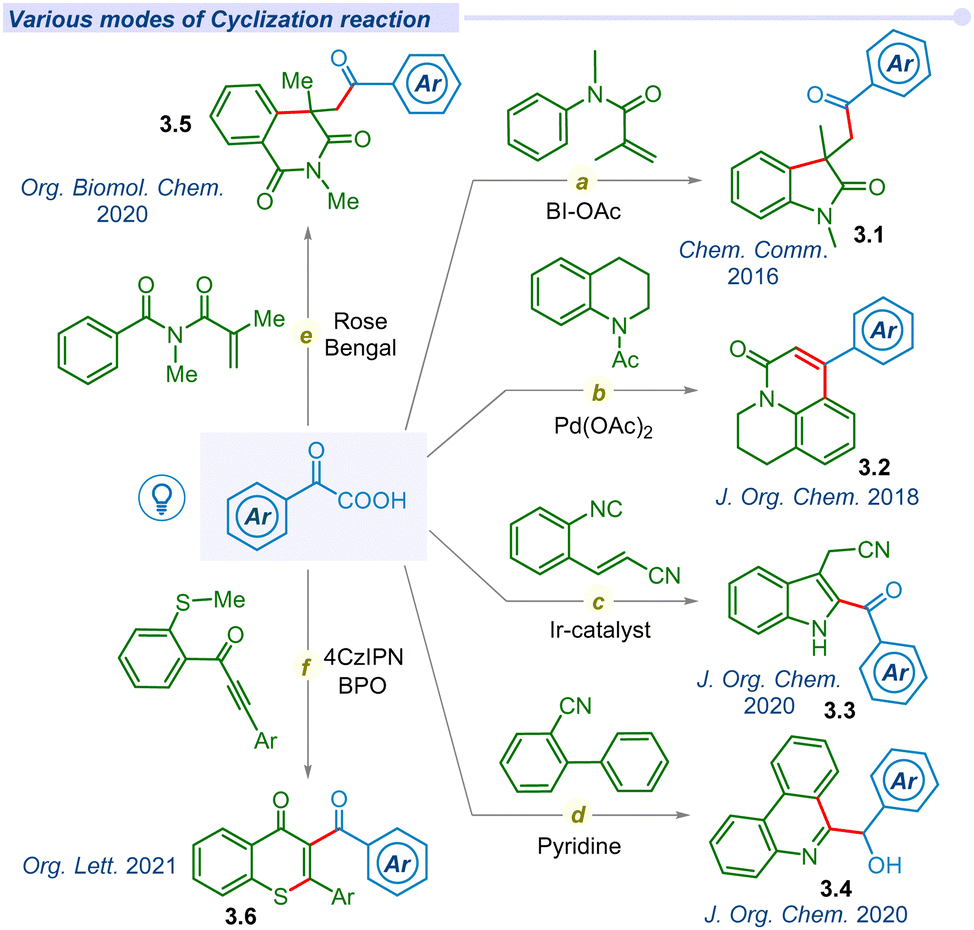 | ||
| Fig. 3 Decarboxylative cyclization of α-keto acids. | ||
2.2 C–hetero cross-coupling and cyclization
In 2014, Lei and co-workers pioneered room temperature visible light-mediated aerobic decarboxylative amidation of α-keto acids with amines using a ruthenium photocatalyst, to unveil new avenues for complex molecular construction via decarboxylation of carboxylic acids (Fig. 4a).30 Later, Xu and co-workers developed a photocatalyst-free singlet oxygen mediated N-acylation (4.1) of amines with α-keto acids.65 | ||
| Fig. 4 Decarboxylative C–hetero cross-coupling and cyclization of α-keto acids. | ||
In 2018, Guo and He groups reported the decarboxylative cyclization of α-keto acids for the production of 2,5-diaryl-1,3,4-oxadiazoles (4.2) using acylhydrazines under visible light conditions (Fig. 4b).66,67 Later on, Sharma and co-workers developed metal and photocatalyst-free cyclization of α-keto acids with 2-amino thiophenol to synthesize benzothiazoles (4.3) under blue LED conditions (Fig. 4c).68 Similarly, the Le group synthesized quinazoline derivatives (4.4) via cyclization of α-keto acids with 2-amino benzylamine under blue LED irradiation (Fig. 4d).69
2.3 Decarboxylation of β-hetero-α-keto acids
The decarboxylation of β-hetero-α-keto acids was achieved in 2013, when Overman and co-workers proposed a novel approach for the exclusive generation of a quaternary carbon center via cross-coupling between Michael acceptors and N-phthalimidoyl oxalate derivatives of tertiary alcohols under photoredox conditions.70 Later in 2014, the MacMillan group reported a redox-neutral approach for quaternary carbon center formation through the cross-coupling of in situ generated alkyl radicals from decarboxylation of carboxylic acids and Michael acceptors with the Ir[dF(CF3)ppy]2(dtbbpy)PF6 photocatalyst.71 In 2015, Overman and MacMillan merged these two features and developed a simple and efficient route for the synthesis of quaternary carbons (5.1) through the cross-coupling of simple oxalate salts of tertiary alcohols with electron-deficient alkenes under photocatalytic conditions (Fig. 5a).72 Thereafter, in 2018 the Landais group reported oxidative decarboxylative generation of a carbamoyl radical from oxamic acids under metal-free conditions.73 They successfully employed the carbamoyl radical to synthesize urethanes and ureas (5.2) from alcohols and amines respectively by using an organo-photocatalyst and HIR (BI-OAc) as an oxidant (Fig. 5b). Similarly, they also coupled this carbamoyl radical into the heteroarenes to afford the corresponding enantiopure amides.74 In the same year, Feng and co-workers utilized this carbamoyl radical for cyclization with electron deficient olefins to unveil an efficient route for the synthesis of 3,4-dihydroquinolin-2(1H)-ones (5.3) (Fig. 5c).75 Mechanistic insight revealed the steps involved in the sequential addition of carbamoyl radical, cyclization, and aromatization in a cascade manner. Recently, the Landais group further coupled this carbamoyl radical into imines for the synthesis of amino acid amides (5.4) under a ferrocene-based photocatalyst (Fig. 5d).76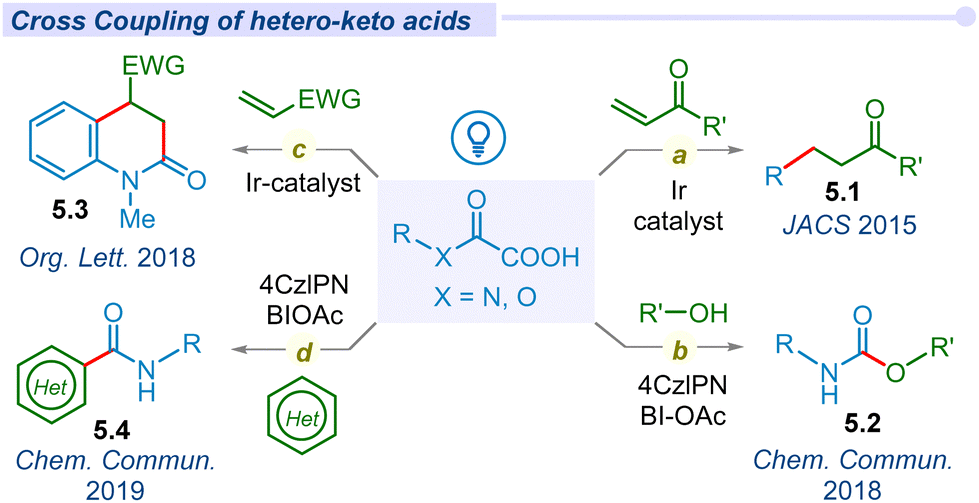 | ||
| Fig. 5 Decarboxylative cross-coupling as well as cyclization of β-hetero-α-keto acids. | ||
3. Decarboxylation of C(sp3)-aliphatic acids
3.1 C(sp3)–Csp2(aromatic) cross-coupling
Developing strategies for C(sp3)–C(sp2) bond formation expands to access a broader chemical space, which is essential for addressing complex synthetic targets such as natural products and potential drug candidates.77 The decarboxylation of aliphatic carboxylic acids has a long-standing history of centuries and is currently undergoing a resurgence through a photocatalytic single electron transfer pathway.Due to the potential application of the C(sp3)–C(sp2) coupling reaction, the MacMillan group pioneered photocatalyzed decarboxylative coupling of α-amino acids with cyanoarene moieties in 2014 (Fig. 6a).78 The irradiation of iridium photocatalyst with a compact fluorescent lamp (CFL) in the presence of CsF as a base, α-amino acids and cyanoarenes afforded the racemic mixture of benzylic amines (6.1). This cross-coupling is also well tolerated for various α-amino and α-oxy acids with electronically diverse cyanoarene derivatives.
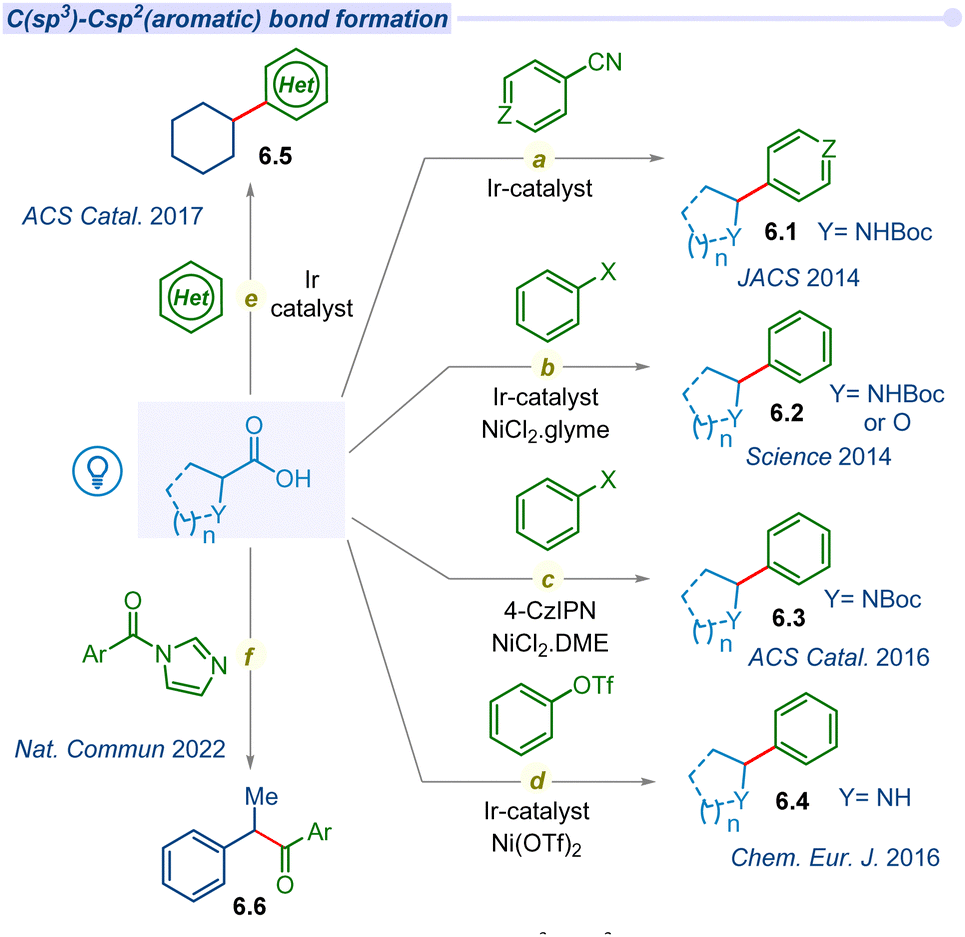 | ||
| Fig. 6 Decarboxylation of aliphatic acids for C(sp3)–Csp2(aromatic) cross-coupling. | ||
In the same year, MacMillan and Doyle significantly expanded the decarboxylative cross-coupling protocol with the groundbreaking approach of merging photoredox with nickel catalysis for a wide range of substituted (hetero)aryl halides in synthetic transformations (Fig. 6b).79 They explored the decarboxylative cross-coupling between amino acids or α-O-carboxylic acids and aryl halides. Subsequently, they developed an asymmetric decarboxylative C(sp3)–C(sp2) cross-coupling under a dual Ir-photocatalyst and a chiral nickel co-catalyst.80 In 2016, the Zhang group developed an alternative and cost-effective organo-fluorophore based photocatalyst (4-CzIPN) to replace the iridium polypyridyl catalyst in merging photoredox/Ni catalytic cross-coupling reactions (Fig. 6c).81 Furthermore, the Rueping group explored the arylation agents for C(sp3)–C(sp2) bond forming decarboxylation reactions beyond aryl halides to aryl triflates in dual iridium/nickel photocatalysis (Fig. 6d).82
Despite the various methods of decarboxylative C–H functionalization of heteroarenes, a visible light-mediated mild and efficient protocol remains elusive. In 2017, the Glorius group reported visible light irradiated direct C–H alkylation of heteroarenes with aliphatic carboxylic acids using iridium photocatalyst and ammonium persulfate as an additive for the first time (Fig. 6e).83 This effective method is used to alkylate a variety of N-heterocycles, including pyridines, isoquinoline, benzothiazole, phthalazine, and other derivatives with cyclic or acyclic carboxylic acids, as well as different kinds of amino acids and fatty acids. A similar type of alkylation of heteroarenes was developed by the Li group using an oxidant-free dual Ir/Co photocatalytic system.84 Thereafter, significant advancements in decarboxylative C(sp3)–C(sp2) coupling with aryl halides using an iridium based photocatalyst and nickel catalyst were reported by several groups. These couplings are also extended towards the synthesis of heteroaryl-C-nucleosides,85 bioconjugation,86,87 and polyfluoroarylation.88,89
Merging of N-heterocyclic carbene (NHC) and photocatalyst based cross-coupling between a radical precursor and an acyl electrophile is emerging as an attractive method for ketone synthesis. However, the previous reports are mostly focused on the synthesis of prefunctionalized radical precursors and their subsequent two-component coupling process. In 2022, Chi and co-workers developed a straightforward method for the synthesis of ketones via cross-coupling of acyl imidazoles and carboxylic acids using the merger of Ir/NHC photocatalysts (Fig. 6f).90 This report also provides a one pot strategy for the synthesis of ketones from two different carboxylic acid derivatives through in situ generated acyl imidazole intermediates. Since then, a myriad of developments on photodecarboxylative C(sp3)–C(sp2) has gained considerable interest within the synthetic community.91–93
3.2 C(sp3)–Csp2(alkene) cross-coupling
Alkenes and their derivatives are among the most fundamental synthetic intermediates and adaptable building blocks, capable of transforming into complex molecular scaffolds with a wide range of applications. Recently, alkenyl C(sp3)–C(sp2) bond-forming reactions utilizing C(sp3)–alkyl carboxylic acids have drawn significant interest in synthetic methodology via a decarboxylative pathway. In 2014, MacMillan introduced a C(sp3)–C(sp2) cross-coupling reaction between N-Boc-α-amino acids with vinyl sulfones (Fig. 7a).94 Under 26 W CFL irradiation this decarboxylative reaction proceeds rapidly in the presence of iridium polypyridyl photocatalyst and CsHCO3 additive. In addition, the same group also reported a decarboxylative vinylation using vinyl halides by altering the coupling partners with alkyl carboxylic acids (Fig. 7b).95 In this reaction, different α-heteroatoms containing alkyl carboxylic acids (α-oxy and α-amino acids) are reacted with vinyl bromides or iodides to produce an important class of allylamine or allylether motifs using dual photoredox/Ni catalysis. | ||
| Fig. 7 Decarboxylation of aliphatic acids for C(sp3)–Csp2(alkenyl) cross-coupling. | ||
In 2017, the Fu group disclosed a similar approach for the vinylation of N-protected α-amino acids using gem-difluoro styrene derivatives (Fig. 7c). All the defluorinated mono-fluoro allylamine products (7.3) with a mixture of (E)- and (Z)-isomers are reported.96 Surprisingly, Shang and co-workers reported a stereo-selective (Z)-olefinated decarboxylative cross-coupled product from styrene derivatives.97
Of late, MacMillan and co-workers developed a new decarboxylative migratory insertion coupling protocol of carboxylic acids with alkynes under visible light irradiation to afford C(sp3)–C(sp2) cross-coupling products 7.4 (Fig. 7d).98
3.3 C(sp3)–C(sp3) cross-coupling
In 2014, MacMillan and co-workers used carboxylic acids as traceless activating groups for radical formation and their 1,4-conjugate addition to a Michael acceptor under visible light-irradiation (Fig. 8a).99 This is the first report of a photodecarboxylative C(sp3)–C(sp3) bond-forming protocol. Most importantly, this newly established approach provides an alternate route to produce Michael donors that does not require organometallic activation or propagation. Lately, the same group developed a highly efficient challenging methodology to form a C(sp3)–C(sp3) bond using an unactivated alkyl halide coupling partner under Ir/Ni merging catalysis (Fig. 8b).100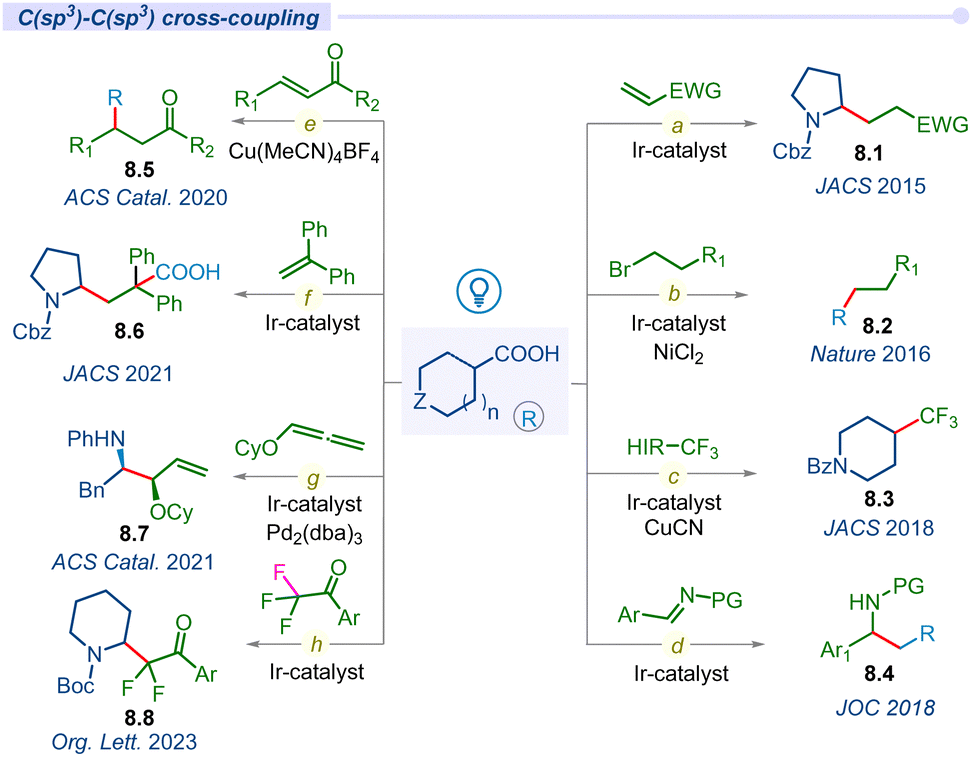 | ||
| Fig. 8 Decarboxylation of aliphatic acids for C(sp3)–C(sp3) cross-coupling. | ||
The incorporation of CF3 groups can enhance therapeutic efficacy by modifying protein–ligand interactions and enhancing membrane permeance. Although significant advancements have been developed for accessing aryl-CF3 motifs, aliphatic C(sp3)–CF3 bond forming methods remain elusive. In this regard, in 2018, the MacMillan group disclosed decarboxylative trifluoromethylation of aliphatic acids using a merging Ir/Cu-system and Togni's reagent as a source of CF3 (Fig. 8c).101
In the same year, the Lu group developed a decarboxylative photo-irradiated benzylation of imines using arylacetic acids (Fig. 8d).102 This reaction tolerates a variety of functional groups, showcasing a broad substrate scope of primary, secondary, and tertiary aliphatic acid precursors. Similarly, they also developed a decarboxylative conjugate addition reaction of carboxylic acids with para-quinone methides (p-QMs) using an organo-photoredox catalyst to synthesize 1,1,2-triarylethanes.103 Subsequently, Larionov and other groups developed direct decarboxylative conjugate addition of a variety of carboxylic acids with different Michael acceptors under visible light conditions for the construction of diverse C(sp3)–C(sp3) bond forming methodologies (Fig. 8e).104–106
For the first time, Yu and co-workers introduced carboxylic acids as bifunctional reagents for a redox-neutral carbo-carboxylation reaction of activated alkenes under photoredox catalysis (Fig. 8f).107 This study represents a unique approach for the usage of both carboxyl and alkyl segments of carboxylic acids simultaneously. In 2021, the Breit group disclosed an asymmetric merging Ir/Pd-catalyzed decarboxylative hydroaminoalkylation of alkoxylallenes for accessing regio- and enantio-selective vinyl 1,2-amino ether products 8.7 (Fig. 8g).108 Recently, the Xing group developed a simultaneous decarboxylation and defluorination approach to achieve difluoroalkylation in amino acids by using readily available amino acids and trifluoroacetophenones as key starting materials (Fig. 8h).109
3.4 C(sp3)–C(sp) cross-coupling
In recent years, there has been a lot of interest in using photoredox catalysis with hypervalent iodine reagents in visible light-mediated synthetic transformation to decarboxylate carboxylic acids. In 2015, Xiao and co-workers developed unprecedented photocatalytic direct radical decarboxylative alkynylation of carboxylic acids under visible-light irradiation (Fig. 9a).110 This methodology efficiently produced various types of alkynes (9.1) and ynone using iridium photocatalyst and ethynylbenziodoxolones (EBX) as the alkynylating agent. Similarly, the Cheng group also developed a metal-free photocatalytic method for decarboxylative alkynylation of carboxylic acids using 9,10-dicyanoanthracene as a photoredox catalyst.111 | ||
| Fig. 9 Decarboxylation of aliphatic acids for C(sp3)–C(sp) cross-coupling. | ||
In 2017, Waser and co-workers developed a one-step transformation of aliphatic acids to their corresponding nitriles under visible light mediated merging Ir/HIR-reagent cyanobenziodoxolones (CBX) (Fig. 9b).112 This approach potentially cyanated a wide variety of amino acids. Later, a similar type of transformation was discovered by the Gomez group in 2019, using a less expensive organo-photocatalyst riboflavin tetraacetate.113
Messaoudi and co-workers developed the dual photoredox/copper catalytic alkynylation approach for the synthesis of anomeric furanosyls using various terminal alkynes (Fig. 9c).114 This reaction offers an extraordinary output for alkynyl C-nucleosides with high efficiency and diastereo-selectivity under mild conditions. In 2022, Zhang and co-workers further reported a sustainable protocol for the synthesis of alkynyl C-glycosides (9.4) with high stereoselectivity with broad substrate scope and good functional group compatibility (Fig. 9d).115
3.5 C(sp3)–halo/hetero cross-coupling
Fluorinated compounds are important structural motifs widely found in pharmaceuticals, agrochemicals, and radiochemical applications. In this regard, Paquin and co-workers developed the pioneering photocatalytic C–F bond formation of aryloxyacetic derivatives using a Selectfluor and ruthenium photocatalyst in 2015.116 Furthermore, the MacMillan group developed a highly regiospecific, bond strength-independent decarboxylative fluorination of aliphatic carboxylic acids using the iridium photocatalyst (Fig. 10a).117 This operationally simple fluorination reaction involved formation of carboxyl radicals from carboxylic acids and the addition to F• from the fluorinating reagent affording the desired fluoroalkane products 10.1.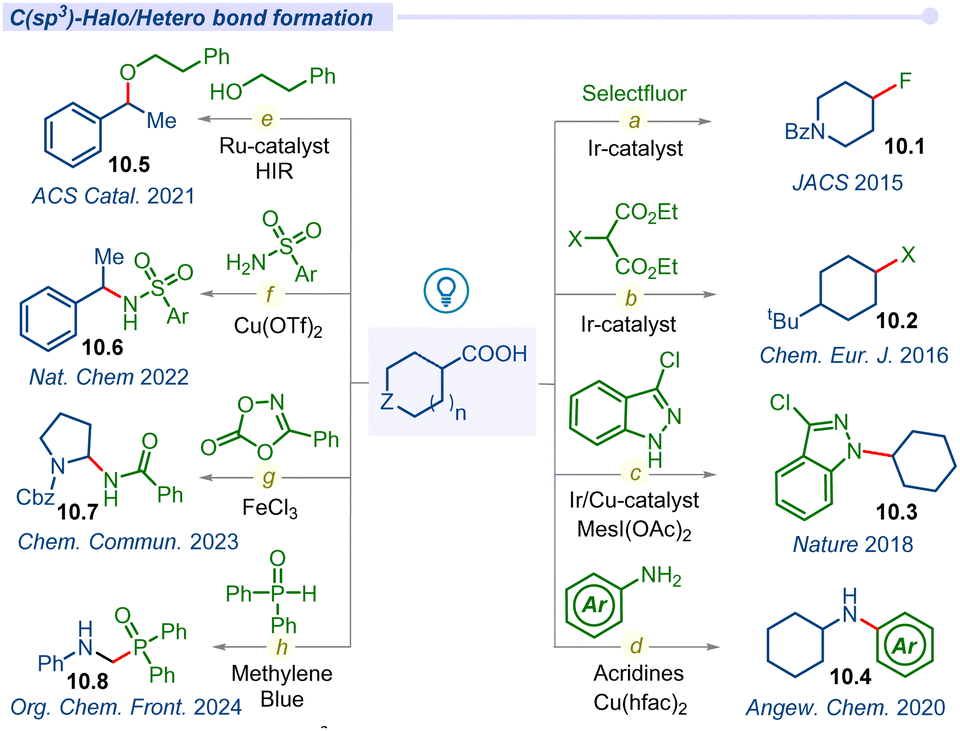 | ||
| Fig. 10 Decarboxylative C(sp3)–halo/hetero cross-coupling of aliphatic carboxylic acids. | ||
Alkyl halides are commonly used substrates in electrophilic substitution and cross-coupling reactions. Due to the synthetic utility of alkyl halides, the Glorius group developed a simple decarboxylative halogenation (chlorination, bromination and iodination) of aliphatic carboxylic acids by employing an iridium photocatalyst (Fig. 10b).118 Afterwards, a similar type of halide transformation was developed by Hu and co-workers by introducing an iron salt as a photocatalyst.119
Nitrogen-containing organic scaffolds are commonly found in natural products, pharmaceuticals, dyes and functional materials. The Buchwald–Hartwig coupling reaction is one of the most traditional reactions for the construction of a C–N bond but suffers from its elevated temperature.120 In 2016, Guan and co-workers disclosed a room temperature photo-induced amination of indoline-2-carboxylic acids and azodicarboxylate ester using metal-free Rose Bengal as a photocatalyst.121 Significant progress has been observed in the construction of diverse C(sp2)–N bonds using a variety of transition metal catalysis strategies but the formation of C(sp3)–N bonds remains the most daunting task in the field of cross-coupling chemistry. In 2018, MacMillan and co-workers addressed this challenge of the C(sp3)–N bond via decarboxylative cross-coupling of naturally abundant alkyl carboxylic acids and feedstock nitrogen nucleophiles as coupling partners under the merging Ir/Cu-photocatalysis pathway (Fig. 10c).122 Lately, in 2020, Larionov and co-workers described a visible-light-enabled N-alkylation of various amines including heterocyclic amines and carboxylic acids using acridine as a photocatalyst with a copper co-catalyst (Fig. 10d).123 They also developed a direct access to sulfonamides and sulfonyl azides from carboxylic acids under visible light conditions.124
Furthermore, the Leonori group also disclosed another visible light induced decarboxylative azidation reaction of cyclic α-amino acids using the Rhodamine 6G organo-photocatalytic pathway.125 In the report, they successfully synthesized new α-N-Boc-amino-azide building blocks and also selectively modified the N-terminal proline residues of dipeptides.
In 2021, Terrett and co-workers used a dual Ru-photoredox/iodine(III) platform to form carbon–oxygen bonds between simple alcohols and carboxylic acids (Fig. 10e).126 They overcame the electronically mismatched nature of previous radical-based decarboxylative couplings. Next year, the Yoon group developed a ligand-to-metal charge transfer (LMCT) decarboxylative nucleophilic cross-coupling reaction of carboxylic acids with various carbon, nitrogen and oxygen nucleophiles under visible-light irradiation (Fig. 10f).127 In mechanistic investigation, they proposed a photoactive Cu(II) carboxylate as a chromophore which formed in situ via interaction between Cu(OTf)2 and a carboxylic acid. Photoexcitation of the LMCT state creates a dissociative carboxyl radical, which can readily decarboxylate to form the corresponding alkyl radical species that facilitated the cross-coupling process.
The Bao group developed an efficient method for the synthesis of amide derivatives via C–N cross-coupling between α-amino acids and dioxazolones (Fig. 10g).128 This method effectively converted α-amino acids to their corresponding amides (10.7) using iron(III) chloride under visible light irradiation.
Most recently Yang and co-workers introduced a simple method for α-amino phosphine oxides via decarboxylative cross-coupling of N-aryl glycines with diarylphosphine oxides using methylene blue (MB) as a photocatalyst (Fig. 10h).129 Similarly, different kinds of decarboxylative C–S/C–Se cross-coupling have also been explored by several groups in the recent past.130–133
3.6 Cyclization of aliphatic acids
Decarboxylative cyclization through multiple C–C and C–Het bonds forming concerted processes manifold a wide range of biologically and pharmaceutically important organic scaffolds. In this perspective, the Studer group in 2017 introduced readily accessible and highly efficient α-imino-oxy propionic acid precursors to synthesize pyrroline derivatives (11.1) using Michael acceptors through SET reduction (Fig. 11a).134 An iridium based photocatalyst accomplished this cascade reaction through sequential intra/inter-molecular C–N and C–C bond formation under mild conditions with a wide range of functional groups tolerance. Subsequently, the Zhou group also reported diastereo- and regio-selective fluorinated benzo[a]quinolizidines (11.2) by the combination of dihydroisoquinoline acetic acids and trifluoromethyl alkenes under visible light conditions (Fig. 11b).135 This decarboxylative cyclization reaction proceeded through a one-pot sequential defluorinative cross-coupling and intramolecular C–H functionalization pathway.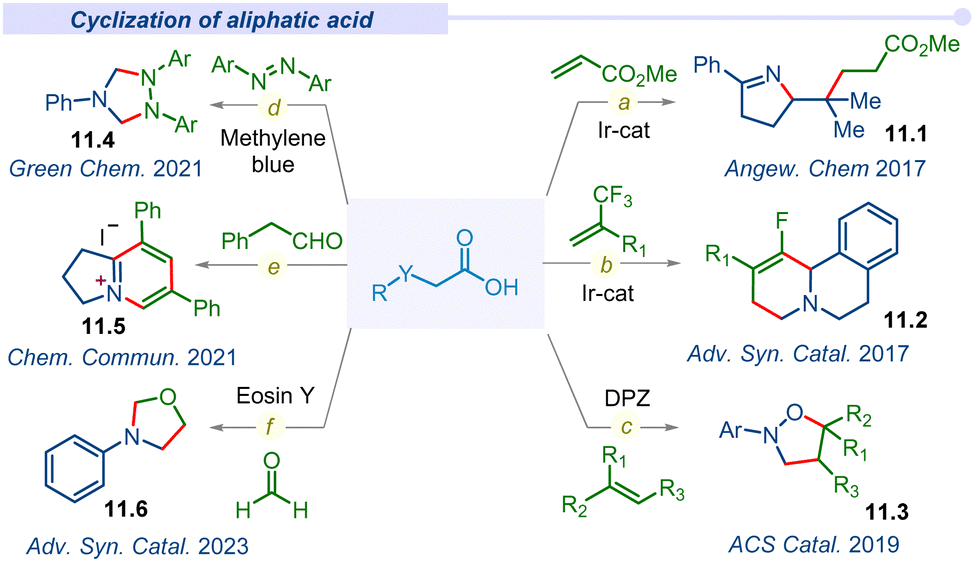 | ||
| Fig. 11 Decarboxylative cyclization of aliphatic carboxylic acids. | ||
In 2019, the Jiang group developed a unique route for the synthesis of pharmaceutically important and bioactive isoxazolidine derivatives (11.3) from naturally abundant amino acids (Fig. 11c).136 This photoinduced transition-metal free cyclization followed concerted difunctionalization of activated alkenes with the decarboxylative α-amino radicals of amino acids. This is the first report of isoxazolines dismissing the traditional 1,3-dipolar or hydroxylamine cyclization route and emerges as an alternative method for a heterocyclic scaffold under an environmentally friendly and atom efficient pathway.
In 2021, the Wang group developed a visible-light-driven cyclization strategy of N-aryl glycines with azobenzenes through a decarboxylation pathway for the formation of 1,2,4-triaryl-1,2,4-triazolidines (11.4) (Fig. 11d).137 Commercially available organic photocatalyst methylene blue was irradiated under visible-light and afforded room temperature triazolidine products with excellent yields. Apart from the previously described metal-catalyzed activation/cascade cyclization via ortho C–H bond activation is the first metal-free example of direct N-heterocycle synthesis by the addition and cyclization of the azo bond of azobenzenes.
Furthermore, the Xia group demonstrated a method for the synthesis of chichibabin pyridinium (11.5) via cross-coupling between α-amino acids and aldehydes under visible light irradiation (Fig. 11e).138 The oxidative decarboxylation of in situ generated electron-rich enamine and sequential new bond formation afforded the desired chichibabin product. Recently in 2023, a complementary approach to acquire oxazolidine derivatives was developed by the Singh group under a visible light irradiated tandem decarboxylation–cyclization plethora using organophotocatalysts.139 Furthermore, subsequent development of various visible light mediated decarboxylative cyclizations was observed by several groups with broad and functional group tolerance.140–144
4. Decarboxylation of α,β-unsaturated acids
α,β-unsaturated acids or acrylic acids are important feedstock chemicals found in Nature in various forms. In particular, 3-phenylacrylic acids, popularly known as cinnamic acids, have drawn lots of attention from organic chemists in the past decades.145,146 The versatility of such moiety to transform into alkyl, β-styryl, β-keto, and α,β-diketo fragments has been employed to develop many underexplored catalytic methods under blue LED conditions. The preparation, availability, thermal stability and natural abundance make this carboxylic acid one of the most explored chemical architectures for decarboxylation to access new C–C/Het bonds under mild and eco-benign conditions.4.1 C–C cross-coupling
In 2014, hypervalent iodine mediated first decarboxylative trifluoromethylation of α,β-unsaturated acids was developed by the Zhu group using Togni's reagent to access fluoromethylated alkene moieties (12.1) under fac-Ir(ppy)3 photocatalytic conditions (Fig. 12a).32 The following year, Chen and co-workers also developed hypervalent acetoxybenziodoxole (BI-OAc) induced chemo-selective C(sp3)–C(sp2) coupling of α,β-unsaturated acids with alkyl trifluoroborate using a ruthenium photocatalyst.147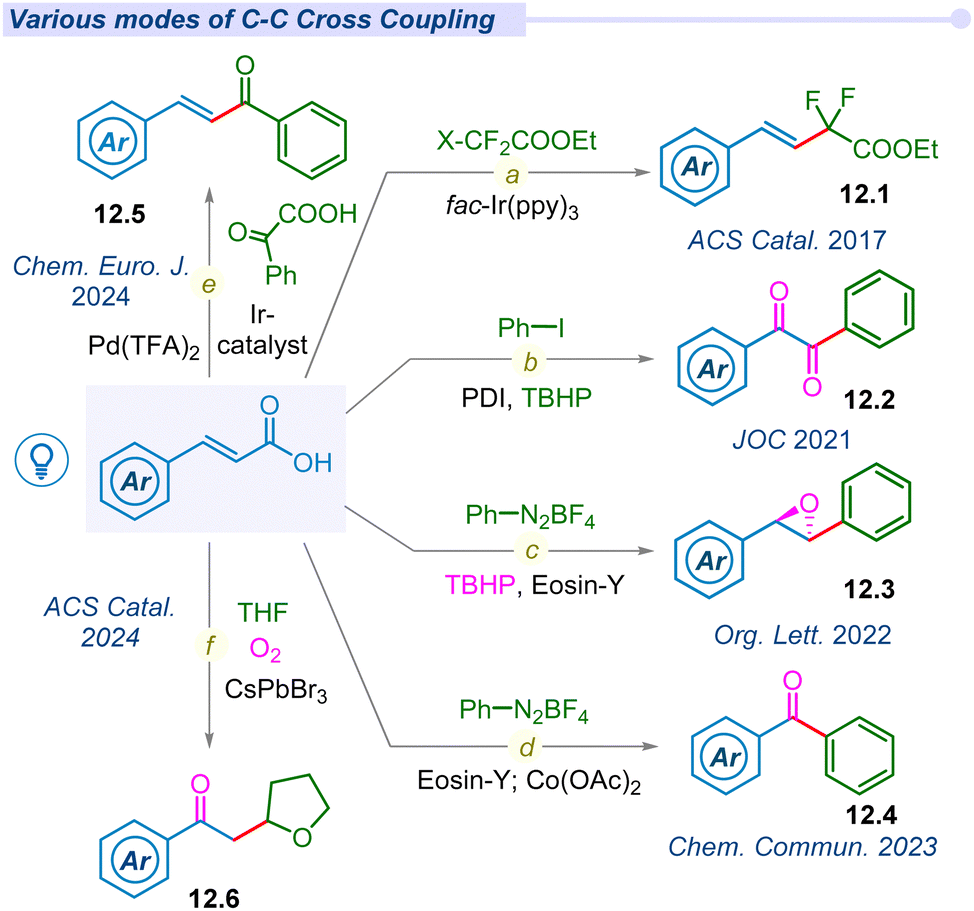 | ||
| Fig. 12 Decarboxylative C–C cross-coupling of unsaturated acids. | ||
In 2017, Timothy Noel and his group developed a novel method to access difluoromethylated styrenes (12.1) using fac-Ir(ppy)3 and ethyl bromodifluoroacetate in both batch and continuous flow processes (Fig. 12a).148 Notably, β-styryl derivatives were synthesized by other groups under similar photocatalytic conditions.149–152 Later, Singh et al. used a metal-free perylenebisimide(PDI) photocatalyst to synthesize benzil derivatives (12.2) from cinnamic acids using iodobenzene and peroxide (Fig. 12b).153 The same group synthesized highly strained stereospecific epoxides (12.3) using diazonium salts, Eosin-Y and peroxide under mild photocatalytic conditions (Fig. 12c).154 Furthermore, they also developed a synthetic route for benzophenone derivatives (12.4) using diazonium salt under cobalt mediated photo-conditions (Fig. 12d).155 Most recently, our group developed dual decarboxylative chemo-selective C(sp3)–C(sp2) coupling from α,β-unsaturated acids and glyoxalic acid under an iridium–palladium dual catalytic system to access (E)-chalcones 12.5 (Fig. 12e).156 In 2024, Datta, Pradhan and Ghosh groups jointly pioneered a dodecahedron shaped heterogeneous photocatalyst CsPbBr3 to access 2-oxyalkylated ketones (12.6) from cinnamic acid and cyclic ethers under transition metal-free conditions (Fig. 12f).157 This report explored the semiconductor–metal heterostructures of CsPbBr3 nanocrystals and their facets dependent photocatalytic behaviour for selective C(sp3)–C(sp3) cross-coupled products.
4.2 C–hetero cross-coupling
Apart from C–C cross-coupling, various modes of decarboxylative C–Het coupling have been reported in recent years. In 2016, the Weng group established α,β-unsaturated acids as β-styryl synthons with sulfonylhydrazides to access vinyl sulfones under metal-free conditions (Fig. 13).158–162 Lately, in 2020, Singh used KSCN as a source of ·SCN to access (E)-vinylthiocyanate derivatives (Fig. 13).163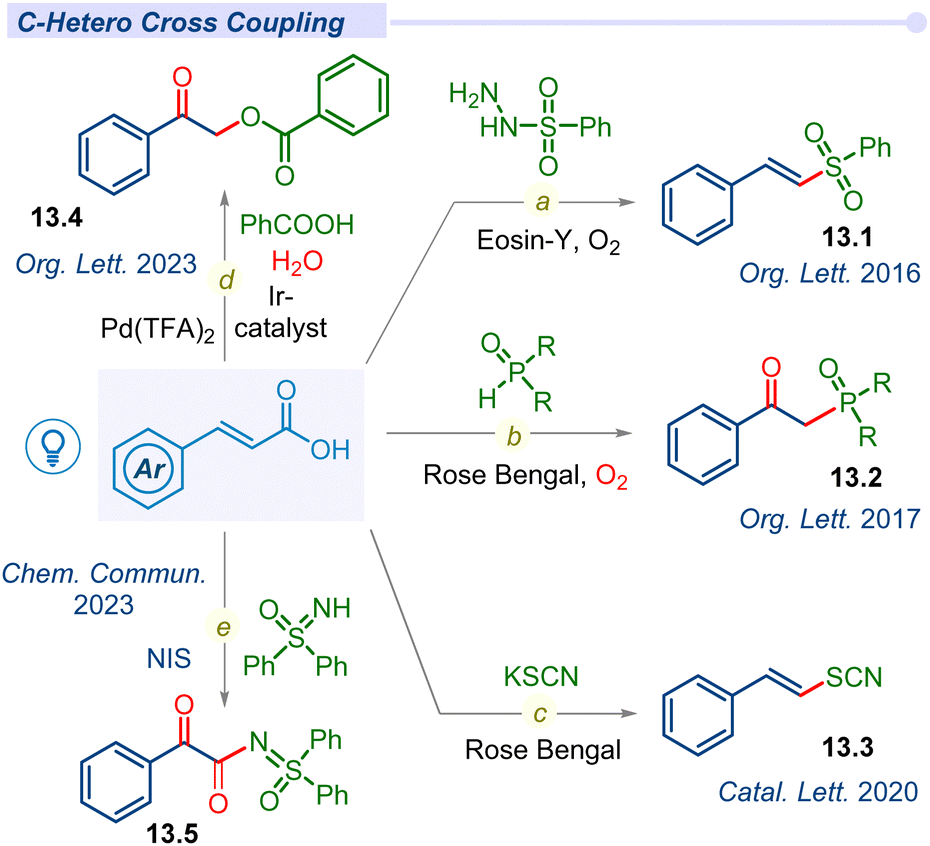 | ||
| Fig. 13 Decarboxylative C–hetero cross-coupling of unsaturated acids. | ||
Surprisingly, other crucial heteroatoms such as phosphorus, oxygen, and nitrogen mostly oxidize such alkene bonds and afford the β-keto or α,β-diketo system under mild conditions. In 2017, the Zou group reported decarboxylative C–P bond forming β-keto-phosphine oxide (13.3) from diarylphosphine oxides with Rose Bengal using a CFL bulb under an oxygen atmosphere (Fig. 13c).164 In this report, a mechanistic investigation revealed that aerial oxygen is oxidizing alkene bonds to access β-keto analogues.
Recently, our group developed decarboxylative C–O cross-coupling of benzoic acids with unsaturated acids to afford the corresponding O-alkylated ester product (13.4) under dual iridium–palladium merging photocatalytic conditions (Fig. 13d).165 Mechanistic investigation revealed that the ketone oxygen source was molecular water whereas aerial oxygen acted as an oxidant in this green and sustainable methodology. Patel and co-workers developed an NIS-initiated oxidative decarboxylative sulfoximidation (13.5) reaction (Fig. 13e).166 This C–N cross-coupling process under blue LED facilitates oxidation upon the alkene bond for dual C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O formation in the desired product. Mechanistic investigation revealed that the source of β-keto oxygen is molecular oxygen and other oxygens are contributed by water molecules.
O formation in the desired product. Mechanistic investigation revealed that the source of β-keto oxygen is molecular oxygen and other oxygens are contributed by water molecules.
5. Decarboxylation of aromatic acids
Over the centuries, enormous efforts have been committed to the functionalization of aromatic carboxylic acids for the construction of diverse carbon–carbon (C–C) and carbon–hetero (C–X) bonds via decarboxylative processes.167–175 Usually, aromatic acid decarboxylation leads to an unstable aryl radical intermediate and its CO2 extrusion requires higher activation energy due to the partial double bond character of the C(Ar)–C(CO2) bond. Therefore, decarboxylation of aromatic carboxylic acid requires unusual photocatalytic conditions.5.1 Photo-induced decarboxylation of aromatic acids
In 2017, the Glorius group first reported visible light mediated photoredox catalysis for decarboxylative aryl radical formation using dimethyl 2-bromo-2-methylmalonate oxidant through an in situ hypobromite intermediate under mild thermal conditions to afford biaryl motifs (14.1) (Fig. 14a).176 Following this pioneering report, photo-decarboxylative iodination of aromatic carboxylic acids was developed by the Fu group under mild conditions using stoichiometric amounts of oxidants.177 Seminal work on thermally photoinduced benzoic acid decarboxylation reactions with electron deficient alkenes, diboranes, and acetonitrile using organic photocatalysts biphenyl (BP) and 9,10-dicyanoanthracene (DCA) or 1,4-dicyanonaphthalene (DCN) was reported by Yoshimi and co-workers in 2020 (Fig. 14b).178 The yield of resulting alkylated adducts, arylboronate esters and protodecarboxylative products was enhanced upon visible light irradiation instead of initial sunlight irradiation.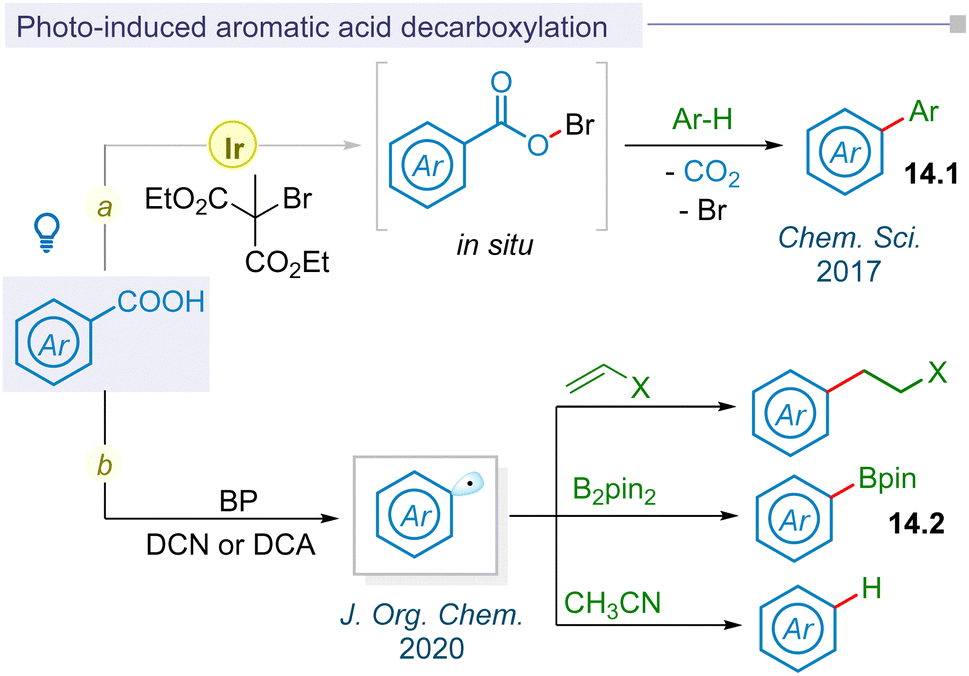 | ||
| Fig. 14 Photoinduced C–C and C–hetero bond formation of aromatic acids. | ||
5.2 LMCT mediated decarboxylation of aromatic acids
In contrast to exogenous reductants or oxidants in Ru or Ir-based polypyridyl photoredox catalysis, regulating the redox state of the metal in photoinduced metal–ligand complexes via ligand-to-metal charge transfer (LMCT) represents a significant advancement in photocatalytic research without the need for a photocatalyst. In 2016, the Doyle group pioneered photocatalytic LMCT for chlorine radical formation to functionalize the inert C(sp3)–H bond.179 Similarly, Wu and Zho groups independently utilized this LMCT strategy to catalyze the formation of chlorine and alkoxy radical for hydroalkylation of alkynes and distal functionalization of primary alcohols.180,181Following this development, the Ritter group reported for the first time LMCT-enabled radical decarboxylation of aromatic acids in 2021 (Fig. 15a).34 This strategy of low-barrier decarboxylative carbometallation proceeded via a highly reactive aryl-copper(III) complex. This complex underwent reductive elimination, thereby regenerating the copper(I) pre-catalyst, along with decarboxylative fluorination (15.1) of the aryl carboxylic acids. In the same year, Ritter reported the decarboxylative hydroxylation of benzoic acids for synthesizing phenols (15.2) (Fig. 15b).182 This method utilized photoinduced LMCT to facilitate charge transfer, initiating the transformation from C–F bonds to C–O bonds. In this LMCT-based decarboxylative functionalization of aromatic carboxylic acids, an excess amount of [Cu(MeCN)4]BF4 and Cu(OTf)2 salts were typically employed.
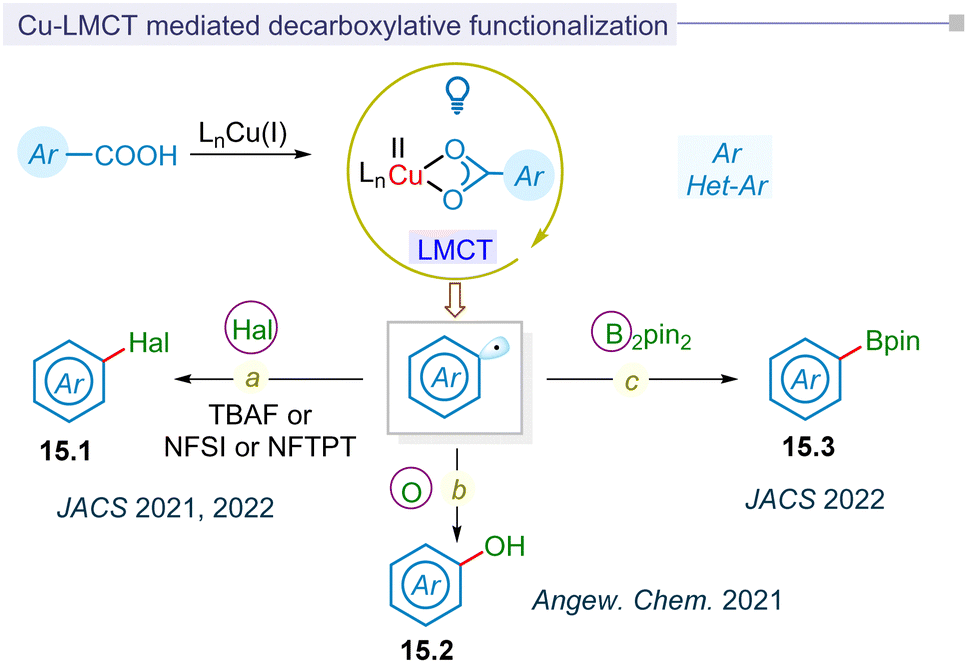 | ||
| Fig. 15 MLCT based C–C and C–hetero bond formation of aromatic acids. | ||
The MacMillan group reported the first catalytic photoinduced LMCT-strategy for synthesizing (hetero)arylboronic esters (15.3) from (hetero)aryl carboxylic acids in 2022 (Fig. 15c).183 The report highlighted a catalytic process involving [Cu(MeCN)4]BF4 that facilitated borylation at ambient temperature. This process was extended to one-pot reactions, including arylation, vinylation, and alkylation by integrating ligand-to-metal charge transfer (LMCT)-borylation with Suzuki–Miyaura cross-coupling. The method demonstrated broad applicability to (hetero)aryl and pharmaceutical substrates. In a similar vein, the decarboxylative halogenation of (hetero)aryl carboxylic acid precursors via a catalytic LMCT-concept was pioneered by the MacMillan group, showcasing a broad substrate scope (Fig. 15a).184 Mechanistically, the process involved the reductive elimination of an aryl-copper(III) intermediate, resulting in fluorination or chlorination. Additionally, an atom transfer pathway facilitated the bromination or iodination of (hetero)aryl carboxylic acids.
5.3 Guanidine based decarboxylative borylation
Photodecarboxylative aryl radical formation and its functionalization have been limited to halogenation, hydroxylation, and borylation, typically achieved through thermal photoredox catalysis or the LMCT concept. Among the various methods for photo-decarboxylative aryl radical formation, biomimetic decarboxylation was most recently reported by the Liu group. This innovative approach utilized a guanidine-based transition state (16.1) to achieve the decarboxylative borylation process through visible-light catalysis (Fig. 16).185 This biomimetic merging of iridium and cobalt catalysis improved the kinetics of decarboxylation and resulted in the borylation (16.2) for carbocyclic and medicinal substrates.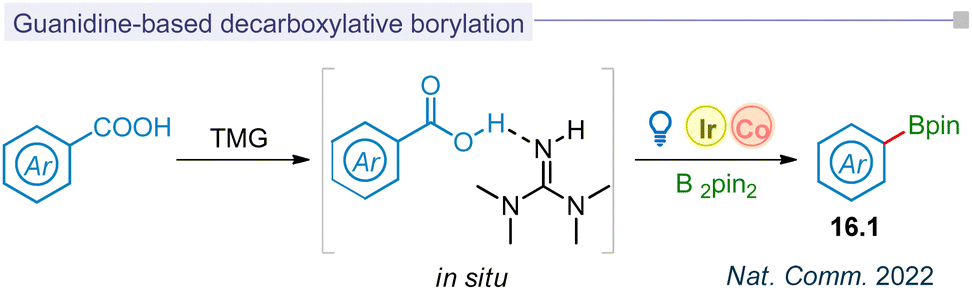 | ||
| Fig. 16 Guanidine based decarboxylative borylation of aromatic acids. | ||
Summary and outlook
As discussed, photo-decarboxylative functionalization of carboxylic acids has gained significant attention due to its diverse applications in various fields with traceless CO2 by-products. In this highlight article, we summarize the complete development of photocatalytic decarboxylation via a concerted single electron transfer (SET) or energy transfer (ET) pathway. This article systematically compiles photodecarboxylative C–C, C–N, C–O, C–S, and C–P cross couplings, along with intra/inter-molecular cyclization of α-keto acids; cyclic and acyclic aliphatic acids; α,β-unsaturated acids and aromatic acids. As decarboxylation is a promising synthetic toolkit for underexplored functionalization/cyclization of various organic molecules, it can easily leverage the molecular library of bio-active scaffolds. A recent upsurge of photomediated protocols has successfully revealed that it can be used for the regio-selective C–H functionalization and cross-coupling. In recent times, considerable progress and rapid access to decarboxylative cross-coupled products disclosed some of the underexplored corners of organic methodology. Although there are numerous reports on C–C cross-coupling, C–Het bond formation is limited for most of the carboxylic acids. Notably, various heteroatoms can influence a reaction for new modes of activation and cyclization in bond-forming methodologies. Interestingly, aryl carboxylic acids are difficult to harness at room temperature and thus LMCT has been employed for decarboxylative functionalization through a lower energy barrier transition state. We hope this article will help the readers gain an insight into the photodecarboxylative functionalization of carboxylic acids over the years.Data availability
This Feature article does not contain any new data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M., S. M. and S. M. acknowledge the Council of Scientific & Industrial Research (CSIR) New Delhi for fellowships. S. P. M. gratefully thanks DST, India for funding (File No.: DST/INSPIRE/04/2022/003369). P. G. acknowledges SERB for the J. C. Bose National Fellowship (JCB/2021/000032).Notes and references
- N. Rodrıguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- T. Patra and D. Maiti, Chem. - Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412–484 CrossRef CAS PubMed.
- J. M. Lowenstein, Citric Acid Cycle, in: Methods in Enzymology, Academic Press, Boston, 1969, vol. 13 Search PubMed.
- H. A. Krebs and P. D. J. Weitman, Krebs’ citric acid cycle: half a century and still turning, Biochemical Society, London, 1987 Search PubMed.
- J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2010, 111, 1846–1913 CrossRef PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- J. Utley, Chem. Soc. Rev., 1997, 3, 157–167 RSC.
- G. J. P. Perry and I. Larrosa, Eur. J. Org. Chem., 2017, 3517–3527 CrossRef CAS PubMed.
- C. V. Wilson, Org. React., 1957, 9, 332 Search PubMed.
- A. F. Shepard, N. R. Winslow and J. R. Johnson, J. Am. Chem. Soc., 1930, 52, 2083–2090 CrossRef CAS.
- M. Nilsson, Acta Chem. Scand., 1966, 20, 423–426 CrossRef CAS.
- A. G. Myers, D. Tanaka and M. R. Mannion, J. Am. Chem. Soc., 2002, 124, 11250–11251 CrossRef CAS PubMed.
- L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef CAS PubMed.
- P. J. Pedersen, D. C. Blakemore, G. M. Chinigo, T. Knauber and D. W. C. MacMillan, J. Am. Chem. Soc., 2023, 145, 21189–21196 CrossRef CAS PubMed.
- M. P. Wiesenfeldt, J. A. Rossi-Ashton, I. B. Perry, J. Diesel, O. L. Garry, F. Bartels, S. C. Coote, X. Ma, C. S. Yeung, D. J. Bennett and D. W. C. MacMillan, Nature, 2023, 618, 513–518 CrossRef CAS PubMed.
- A. V. Tsymbal, L. D. Bizzini and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 21278–21286 CrossRef CAS PubMed.
- R. Shang and L. Lu, Sci. China: Chem., 2011, 54, 1670–1687 CrossRef CAS.
- C. L. Joe and A. G. Doyle, Angew. Chem., Int. Ed., 2016, 55, 4040–4043 CrossRef CAS PubMed.
- S. Maiti, P. Ghosh, D. Raja, S. Ghosh, S. Chatterjee, V. Sankar, S. Roy, G. K. Lahiri and D. Maiti, Nat. Catal., 2024, 7, 285–294 CrossRef CAS.
- P. R. D. Murray, I. N.-M. Liebler, S. M. Hell, E. Villalona, A. G. Doyle and R. R. Knowles, ACS Catal., 2022, 12, 13732–13740 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- Z. Dong and D. W. C. MacMillan, Nature, 2021, 598, 451–456 CrossRef CAS PubMed.
- W. Liu, M. N. Lavagnino, C. A. Gould, J. Alcázar and D. W. C. MacMillan, Science, 2021, 374, 1258–1263 CrossRef CAS PubMed.
- J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed.
- Y. Jin and H. Fu, Asian J. Org. Chem., 2017, 6, 368–385 CrossRef CAS.
- M. Rahman, A. Mukherjee, I. S. Kovalev, D. S. Kopchuk, G. V. Zyryanov, M. V. Tsurkan, A. Majee, B. C. Ranu, V. N. Charushin, O. N. Chupakhin and S. Santra, Adv. Synth. Catal., 2019, 361, 2161–2214 CrossRef CAS.
- Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 7854–7856 RSC.
- J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 502–506 CrossRef CAS PubMed.
- S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef CAS PubMed.
- P. Xu, A. Abdukader, K. Hu, Y. Cheng and C. Zhu, Chem. Commun., 2014, 50, 2308–2310 RSC.
- J. W. Hilborn and J. A. Pincock, J. Am. Chem. Soc., 1991, 113, 2683–2686 CrossRef CAS.
- P. Xu, P. López-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349–5354 CrossRef CAS PubMed.
- C. Anyaegbu, G. Vidali, D. Haridas and J. F. Hooper, Polym. Chem., 2024, 15, 2537–2547 RSC.
- L. J. Gooßen, F. Rudolphi, C. Oppel and N. Rodríguez, Angew. Chem., Int. Ed., 2008, 47, 3043–3045 CrossRef PubMed.
- L.-L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929–7933 CrossRef CAS PubMed.
- W.-M. Cheng, R. Shang, H.-Z. Yu and Y. Fu, Chem. - Eur. J., 2015, 21, 13191–13195 CrossRef CAS PubMed.
- P. Xie, C. Xue, C. Wang, D. Du and S. Shi, Org. Chem. Front., 2021, 8, 3427–3433 RSC.
- M. He, X. Yu, Y. Wang, F. Li and M. Bao, J. Org. Chem., 2021, 86, 5016–5025 CrossRef CAS PubMed.
- D.-L. Zhu, Q. Wu, D. J. Young, H. Wang, Z.-G. Ren and H.-X. Li, Org. Lett., 2020, 22, 6832–6837 CrossRef CAS PubMed.
- B. Zhao, R. Shang, W.-M. Cheng and Y. Fu, Org. Chem. Front., 2018, 5, 1782–1786 RSC.
- H. Huang, G. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 7872–7876 CrossRef CAS PubMed.
- G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830–4833 CrossRef CAS PubMed.
- J.-Q. Chen, R. Chang, Y.-L. Wei, J.-N. Mo, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2018, 83, 253–259 CrossRef CAS PubMed.
- J.-J. Zhao, H.-H. Zhang, X. Shen and S. Yu, Org. Lett., 2019, 21, 913–916 CrossRef CAS PubMed.
- H.-H. Zhang and S. Yu, Org. Lett., 2019, 21, 3711–3715 CrossRef CAS PubMed.
- M. Zhang, J. Xi, R. Ruzi, N. Li, Z. Wu, W. Li and C. Zhu, J. Org. Chem., 2017, 82, 9305–9311 CrossRef CAS PubMed.
- J.-J. Zhang, Y.-B. Cheng and X.-H. Duan, Chin. J. Chem., 2017, 35, 311–315 CrossRef CAS.
- C. Zhou, P. Li, X. Zhu and L. Wang, Org. Lett., 2015, 17, 6198–6201 CrossRef CAS PubMed.
- N. Xu, P. Li, Z. Xie and L. Wang, Chem. - Eur. J., 2016, 22, 2236–2242 CrossRef CAS PubMed.
- L. Gu, C. Jin, J. Liu, H. Zhang, M. Yuan and G. Li, Green Chem., 2016, 18, 1201–1205 RSC.
- Q. Shi, P. Li, X. Zhu and L. Wang, Green Chem., 2016, 18, 4916–4923 RSC.
- Y. Lv, P. Bao, H. Yue, J.-S. Li and W. Wei, Green Chem., 2019, 21, 6051–6055 RSC.
- S. Manna and K. R. Prabhu, J. Org. Chem., 2019, 84, 5067–5077 CrossRef CAS PubMed.
- L.-Y. Xie, Y.-S. Bai, X.-Q. Xu, X. Peng, H.-S. Tang, Y. Huang, Y.-W. Lin, Z. Cao and W.-M. He, Green Chem., 2020, 22, 1720–1725 RSC.
- C. Hou, S. Sun, Z. Liu, H. Zhang, Y. Liu, Q. An, J. Zhao, J. Ma, Z. Sun and W. Chu, Adv. Synth. Catal., 2021, 363, 2806–2812 CrossRef CAS.
- S. Nandi, P. Das, S. Das, S. Mondal and R. Jana, Green Chem., 2023, 25, 3633–3643 RSC.
- W. Ji, H. Tan, M. Wang, P. Li and L. Wang, Chem. Commun., 2016, 52, 1462–1465 RSC.
- C. Wang, J. Qiao, X. Liu, H. Song, Z. Sun and W. Chu, J. Org. Chem., 2018, 83, 1422–1430 CrossRef CAS PubMed.
- X. Zhang, P. Zhu, R. Zhang, X. Li and T. Yao, J. Org. Chem., 2020, 85, 9503–9513 CrossRef CAS PubMed.
- W. Shi, F. Ma, P. Li, L. Wang and T. Miao, J. Org. Chem., 2020, 85, 13808–13817 CrossRef CAS PubMed.
- Y. Su, R. Zhang, W. Xue, X. Liu, Y. Zhao, K.-H. Wang, D. Huang, C. Huo and Y. Hu, Org. Biomol. Chem., 2020, 18, 1940–1948 RSC.
- H.-L. Zhu, F.-L. Zeng, X.-L. Chen, K. Sun, H.-C. Li, X.-Y. Yuan, L.-B. Qu and B. Yu, Org. Lett., 2021, 23, 2976–2980 CrossRef CAS PubMed.
- W.-T. Xu, B. Huang, J.-J. Dai, J. Xu and H.-J. Xu, Org. Lett., 2016, 18, 3114–3117 CrossRef CAS PubMed.
- P. Diao, Y. Ge, W. Zhang, C. Xu, N. Zhang and C. Guo, Tetrahedron Lett., 2018, 59, 767–770 CrossRef CAS.
- L. Wang, Y. Wang, Q. Chen and M. He, Tetrahedron Lett., 2018, 59, 1489–1492 CrossRef CAS.
- A. Monga, S. Bagchi, R. K. Soni and A. Sharma, Adv. Synth. Catal., 2020, 362, 2232–2237 CrossRef.
- C.-H. Hu and Y. Li, J. Org. Chem., 2023, 88, 6401–6406 CrossRef CAS PubMed.
- G. L. Lackner, K. W. Quasdorf and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 15342–15345 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef CAS PubMed.
- G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337–9340 RSC.
- A. H. Jatoi, G. G. Pawar, F. Robert and Y. Landais, Chem. Commun., 2019, 55, 466–469 RSC.
- Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Org. Lett., 2018, 20, 2172–2175 CrossRef CAS PubMed.
- M. Badufle, F. Robert and Y. Landais, RSC Adv., 2024, 14, 12528–12532 RSC.
- A. W. Dombrowski, N. J. Gesmundo, A. L. Aguirre, K. A. Sarris, J. M. Young, A. R. Bogdan, M. C. Martin, S. Gedeon and Y. Wang, ACS Med. Chem. Lett., 2020, 11, 597–604 CrossRef CAS PubMed.
- Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- L. Fan, J. Jia, H. Hou, Q. Lefebvre and M. Rueping, Chem. - Eur. J., 2016, 22, 16437–16440 CrossRef CAS PubMed.
- R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS.
- W.-F. Tian, C.-H. Hu, K.-H. He, X.-Y. He and Y. Li, Org. Lett., 2019, 21, 6930–6935 CrossRef CAS PubMed.
- Y. Ma, S. Liu, Y. Xi, H. Li, K. Yang, Z. Cheng, W. Wang and Y. Zhang, Chem. Commun., 2019, 55, 14657–14660 RSC.
- D. Kölmel, J. Meng, M.-H. Tsai, J. Que, R. P. Loach, T. Knauber, J. Wan and M. E. Flanagan, ACS Comb. Sci., 2019, 21, 588–597 CrossRef PubMed.
- D. K. Kölmel, R. P. Loach, T. Knauber and M. E. Flanagan, ChemMedChem, 2018, 13, 2159–2165 CrossRef PubMed.
- H. Wang, C.-F. Liu, Z. Song, M. Yuan, Y. A. Ho, O. Gutierrez and M. J. Koh, ACS Catal., 2020, 10, 4451–4459 CrossRef CAS.
- X. Sun and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 10557–10562 CrossRef CAS PubMed.
- S. Ren, X. Yang, B. Mondal, C. Mou, W. Tian, Z. Jin and Y. R. Chi, Nat. Commun., 2022, 13, 2846–2855 CrossRef CAS PubMed.
- L. Li, Y. Yao and N. Fu, Eur. J. Org. Chem., 2023, e202300166 CrossRef CAS.
- L. M. Kammer, S. O. Badir, R. M. Hu and G. A. Molander, Chem. Sci., 2021, 12, 5450–5457 RSC.
- J. Guo, D. Norris, A. Ramirez, J. L. Sloane, E. M. Simmons, J. M. Ganley, M. S. Oderinde, T. G. M. Dhar, G. H. M. Davies and T. C. Sherwood, ACS Catal., 2023, 13, 11910–11918 CrossRef CAS.
- A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed.
- A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624–627 CrossRef CAS PubMed.
- J. Li, Q. Lefebvre, H. Yang, Y. Zhao and H. Fu, Chem. Commun., 2017, 53, 10299–10302 RSC.
- C. Zheng, W.-M. Cheng, H.-L. Li, R.-S. Na and R. Shang, Org. Lett., 2018, 20, 2559–2563 CrossRef CAS PubMed.
- N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701–5705 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed.
- J. A. Kautzky, T. Wang, R. W. Evans and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 6522–6526 CrossRef CAS PubMed.
- J. Guo, Q.-L. Wu, Y. Xie, J. Weng and G. Lu, J. Org. Chem., 2018, 83, 12559–12567 CrossRef CAS PubMed.
- J. Guo, G.-B. Huang, Q.-L. Wu, Y. Xie, J. Weng and G. Lu, Org. Chem. Front., 2019, 6, 1955–1960 RSC.
- H. T. Dang, G. C. Haug, V. T. Nguyen, N. T. H. Vuong, V. D. Nguyen, H. D. Arman and O. V. Larionov, ACS Catal., 2020, 10, 11448–11457 CrossRef CAS PubMed.
- Q.-F. Bao, M. Li, Y. Xia, Y.-Z. Wang, Z.-Z. Zhou and Y.-M. Liang, Org. Lett., 2021, 23, 1107–1112 CrossRef CAS PubMed.
- S. Kim, B. Park, G. S. Lee and S. H. Hong, J. Org. Chem., 2023, 88(10), 6532–6537 CrossRef CAS PubMed.
- L.-L. Liao, G.-M. Cao, Y.-X. Jiang, X.-H. Jin, X.-L. Hu, J. J. Chruma, G.-Q. Sun, Y.-Y. Gui and D.-G. Yu, J. Am. Chem. Soc., 2021, 143, 2812–2821 CrossRef CAS PubMed.
- J. Zheng, A. Nikbakht and B. Breit, ACS Catal., 2021, 11, 3343–3350 CrossRef CAS.
- K. Tan, J. He, Z. Mu, I. M. Ammar, C. Che, J. Geng and Q. Xing, Org. Lett., 2023, 25, 8733–8738 CrossRef CAS PubMed.
- Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem. Int. Ed., 2015, 54, 11196–11199 CrossRef CAS PubMed.
- C. Yang, J.-D. Yang, Y.-H. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2016, 81, 12357–12363 CrossRef CAS PubMed.
- F. Le Vaillant, M. D. Wodrich and J. Waser, Chem. Sci., 2017, 8, 1790–1800 RSC.
- N. P. Ramirez, B. König and J. C. Gonzalez-Gomez, Org. Lett., 2019, 21, 1368–1373 CrossRef CAS PubMed.
- M. Zhu and S. Messaoudi, ACS Catal., 2021, 11, 6334–6342 CrossRef CAS.
- K. Lu, Y. Ma, S. Liu, S. Guo and Y. Zhang, Chin. J. Chem., 2022, 40, 681–686 CrossRef CAS.
- M. Rueda-Becerril, O. Mahé, M. Drouin, M. B. Majewski, J. G. West, M. O. Wolf, G. M. Sammis and J.-F. Paquin, J. Am. Chem. Soc., 2014, 136, 2637–2641 CrossRef CAS PubMed.
- S. Ventre, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 5654–5657 CrossRef CAS PubMed.
- L. Candish, E. A. Standley, A. Gómez-Suárez, S. Mukherjee and F. Glorius, Chem. - Eur. J., 2016, 22, 9971–9974 CrossRef CAS PubMed.
- J. Qian, Y. Zhang, W. Zhao and P. Hu, Chem. Commun., 2024, 60, 2764–2767 RSC.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- M.-J. Zhang, G. M. Schroeder, Y.-H. He and Z. Guan, RSC Adv., 2016, 6, 96693–96699 RSC.
- Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–88 CrossRef CAS PubMed.
- V. T. Nguyen, V. D. Nguyen, G. C. Haug, N. T. H. Vuong, H. T. Dang, H. D. Arman and O. V. Larionov, Angew. Chem. Int. Ed., 2020, 59, 7921–7927 CrossRef CAS PubMed.
- V. T. Nguyen, V. D. Nguyen, G. C. Haug, N. T. H. Vuong, H. T. Dang, H. D. Arman and O. V. Larionov, Angew. Chem. Int. Ed., 2020, 59, 7921–7927 CrossRef CAS PubMed.
- D. C. Marcote, R. Street-Jeakings, E. Dauncey, J. J. Douglas, A. Ruffoni and D. Leonori, Org. Biomol. Chem., 2019, 17, 1839–1842 RSC.
- P. Li, J. R. Zbieg and J. A. Terrett, ACS Catal., 2021, 11, 10997–11004 CrossRef CAS.
- Q. Y. Li, S. N. Gockel, G. A. Lutovsky, K. S. DeGlopper, N. J. Baldwin, M. W. Bundesmann, J. W. Tucker, S. W. Bagley and T. P. Yoon, Nat. Chem., 2022, 14, 94–99 CrossRef CAS PubMed.
- Z. Liang, K. Wang, Q. Sun, Y. Peng and X. Bao, Chem. Commun., 2023, 59, 752–755 RSC.
- J. Wen, X. Sun, K. Yan, T. Yan, Z. Liu, Y. Li and J. Yang, Org. Chem. Front., 2024, 11, 796–801 RSC.
- L. Candish, L. Pitzer, A. Gómez-Suárez and F. Glorius, Chem. - Eur. J., 2016, 22, 4753–4756 CrossRef CAS PubMed.
- A.-M. Hu, J.-L. Tu, M. Luo, C. Yang, L. Guo and W. Xia, Org. Chem. Front., 2023, 10, 4764–4773 RSC.
- Y. Gao, R. Hua, H. Yin and F.-X. Chen, Org. Chem. Front., 2023, 10, 2538 RSC.
- J. Vigier, M. Gao, P. Jubault, H. Lebel and T. Besset, Chem. Commun., 2024, 60, 196–199 RSC.
- H. Jiang and A. Studer, Angew. Chem. Int. Ed., 2017, 129, 12441–12444 CrossRef.
- H. Chen, T. Xiao, L. Li, D. Anand, Y. He and L. Zhou, Adv. Synth. Catal., 2017, 359, 3642–3647 CrossRef CAS.
- H. Yang, G. Wei and Z. Jiang, ACS Catal., 2019, 9, 9599–9605 CrossRef CAS.
- J. Yang, M. Song, H. Zhou, Y. Qi, B. Ma and X.-C. Wang, Green Chem., 2021, 23, 5806–5811 RSC.
- Z. Pan, F. Hu, D. Jiang, Y. Liu and C. Xia, Chem. Commun., 2021, 57, 1222–1225 RSC.
- G. Udari, V. Murugesh, N. Sabarinathan, B. Sridhar and S. P. Singh, Adv. Synth. Catal., 2023, 365, 4502–4506 CrossRef CAS.
- S. Pan, M. Jiang, G. Zhong, L. Dai, Y. Zhou, K. Wei and X. Zeng, Org. Chem. Front., 2020, 7, 4043–4049 RSC.
- F. Gao, J.-T. Wang, L.-L. Liu, N. Ma, C. Yang, Y. Gao and W. Xia, Chem. Commun., 2017, 53, 8533–8536 RSC.
- Y. Yu, W. Yuan, H. Huang, Z. Cai, P. Liu and P. Sun, J. Org. Chem., 2018, 83, 1654–1660 CrossRef CAS PubMed.
- Q.-W. Gui, F. Teng, H. Yang, C. Xun, W.-J. Huang, Z.-Q. Lu, M.-X. Zhu, W.-T. Ouyang and W.-M. He, Chem. – Asian J., 2022, 17, e202101139 CrossRef CAS PubMed.
- X. Wang, A. Shi, X.-Q. Huang, X. Chen, T. Li, L. Qua and B. Yu, Org. Biomol. Chem., 2022, 20, 3798–3802 RSC.
- L. Chen, L. Zhang, G. Yan and D. Huang, Asian J. Org. Chem., 2020, 9, 842–862 CrossRef CAS.
- N. Ruwizhi and B. A. Aderibigbe, Int. J. Mol. Sci., 2020, 21(16), 5712 CrossRef CAS PubMed.
- H. Huang, K. Jia and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 1881–1884 CrossRef CAS PubMed.
- X.-J. Wei, W. Boon, V. Hessel and T. Noël, ACS Catal., 2017, 7, 7136–7140 CrossRef CAS PubMed.
- X. Zhu, M.-Y. Han, P. Li and L. Wang, Org. Chem. Front., 2017, 4, 1640–1646 RSC.
- N. Xiong, C. Zhou, S. Li, S. Wang, C. Ke, Z. Rong, Y. Li and R. Zeng, Org. Lett., 2024, 26, 2029–2033 CrossRef CAS PubMed.
- X. Y. Lu, R. Huang, Z. Z. Wang, X. Zhang, F. Jiang, G. X. Yang, F. Y. Shui, M. X. Su, Y. X. Sun and H. L. Sun, J. Org. Chem., 2024, 89, 6494–6505 CrossRef CAS PubMed.
- K. Xu, Z. Tan, H. Zhang, J. Liu, S. Zhang and Z. Wang, Chem. Commun., 2017, 53, 10719–10722 RSC.
- S. Chand, A. K. Pandey, R. Singh and K. N. Singh, J. Org. Chem., 2021, 86, 6486–6493 CrossRef CAS PubMed.
- S. Chand, A. K. Sharma, A. K. Pandey and K. N. Singh, Org. Lett., 2022, 24, 6423–6427 CrossRef CAS PubMed.
- S. Chand, A. K. Sharma, A. K. Pandey and K. N. Singh, Chem. Commun., 2023, 59, 14827–14830 RSC.
- S. Mondal, S. P. Midya, S. Mondal, S. Das and P. Ghosh, Chem. - Eur. J., 2024, 30, e202303337 CrossRef CAS PubMed.
- S. Mondal, S. Banerjee, S. Bera, S. Mondal, S. P. Midya, R. Jana, R. K. Behera, A. Datta, N. Pradhan and P. Ghosh, ACS Catal., 2024, 14, 6633–6643 CrossRef CAS.
- S. Cai, Y. Xu, D. Chen, L. Li, Q. Chen, M. Huang and W. Weng, Org. Lett., 2016, 18, 2990–2993 CrossRef CAS PubMed.
- R. Chawla, S. Jaiswal, P. K. Dutta and L. D. S. Yadav, Tetrahedron Lett., 2020, 61, 151898 CrossRef CAS.
- Q.-Q. Ge, J.-S. Qian and J. Xu, J. Org. Chem., 2019, 84, 8691–8701 CrossRef CAS PubMed.
- X. Li, M. Wang, Z. Wang and L. Wang, Asian J. Org. Chem., 2019, 8, 1426–1435 CrossRef.
- P. Li and G.-W. Wang, Org. Biomol. Chem., 2019, 17, 5578–5585 RSC.
- D. Jaiswal, J. Tiwari, S. Singh, Kartikey, J. Singh and J. Singh, Catal. Lett., 2021, 151, 1738–1744 CrossRef CAS.
- H.-F. Qian, C.-K. Li, Z.-H. Zhou, Z.-K. Tao, A. Shoberu and J.-P. Zou, Org. Lett., 2018, 20, 5947–5951 CrossRef CAS PubMed.
- S. Mondal, S. Mondal, S. P. Midya, S. Das, S. Mondal and P. Ghosh, Org. Lett., 2023, 25, 184–189 CrossRef CAS PubMed.
- N. Chakraborty, K. K. Rajbongshi, A. Dahiya, B. Das, A. Vaishnani and B. K. Patel, Chem. Commun., 2023, 59, 2779–2782 RSC.
- Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed.
- M. P. Drapeau and L. J. Goossen, Chem. - Eur. J., 2013, 22, 18654–18677 CrossRef PubMed.
- S. Bhadra, W. I. Dzik and L. J. Goossen, J. Am. Chem. Soc., 2012, 134, 9938–9941 CrossRef CAS PubMed.
- J. D. Tibbetts, H. E. Askey, Q. Cao, J. D. Grayson, S. L. Hobson., G. D. Johnson, J. C. Turner-Dore and A. J. Cresswell, Synthesis, 2023, 3239–3250 CrossRef CAS.
- S. Katiyar, A. Kumara and K. V. Sashidhara, Chem. Commun., 2022, 58, 7297–7300 RSC.
- L. Zhou, M. Sun, F. Zhou, G. Deng, Y. Yang and Y. Liang, Org. Lett., 2021, 23, 7150–7155 CrossRef CAS PubMed.
- K. Jing, J.-P. Yao, Z.-Y. Li, Q.-L. Li, H.-S. Lin and G.-W. Wang, J. Org. Chem., 2017, 82, 12715–12725 CrossRef CAS PubMed.
- S. U. Dighe, K. S. A. Kumar, S. Srivastava, P. Shukla, S. Singh, M. Dikshit and S. Batra, J. Org. Chem., 2015, 80, 99–108 CrossRef CAS PubMed.
- P. Y. Yeung, K. H. Chung and F. Y. Kwong, Org. Lett., 2011, 13, 2912–2915 CrossRef CAS PubMed.
- L. Candish, M. Freitag, T. Gensch and F. Glorius, Chem. Sci., 2017, 8, 3618–3622 RSC.
- M. Jiang, H. Yang, Y. Jin, L. Ou and H. Fu, Synlett, 2018, 1572–1577 CAS.
- S. Kubosaki, H. Takeuchi, Y. Iwata, Y. Tanaka, K. Osaka, M. Yamawaki, T. Morita and Y. Yoshimi, J. Org. Chem., 2020, 85, 5362–5369 CrossRef CAS PubMed.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- H. P. Deng, X. Z. Fan, Z. H. Chen, Q. H. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579–13584 CrossRef CAS PubMed.
- A. Hu, J. J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef CAS PubMed.
- W. Su, P. Xu and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 24012–24017 CrossRef CAS PubMed.
- N. W. Dow, P. S. Pedersen, T. Q. Chen, D. C. Blakemore, A. Dechert-Schmitt, T. Knauber and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6163–6172 CrossRef CAS PubMed.
- T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A. Dechert-Schmitt, T. Knauber, F. N. Castellano and W. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 8296–8305 CrossRef CAS PubMed.
- Q. Wei, Y. Lee, W. Liang, X. Chen, B. Mu, X. Cui, W. Wu, S. Bai and Z. Liu, Nat. Commun., 2022, 13, 7112 CrossRef CAS PubMed.
Footnote |
| † S. Mondal, S. Mandal and S. Mondal contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |