
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation of a chloride-capped cerium polyoxo nanocluster built from 52 metal ions†
Anamar
Blanes-Díaz
a,
Jennifer N.
Wacker‡
 a,
Jennifer E. S.
Szymanowski
b,
Jeffery A.
Bertke
a and
Karah E.
Knope
*a
a,
Jennifer E. S.
Szymanowski
b,
Jeffery A.
Bertke
a and
Karah E.
Knope
*a
aDepartment of Chemistry, Georgetown University, Washington, D.C. 20057, USA. E-mail: kek44@georgetown.edu
bDepartment of Civil & Environmental Engineering & Earth Sciences, University of Notre Dame, Notre Dame, IN 46556, USA
First published on 15th August 2024
Abstract
Four cerium compounds – (HPy)2[CeCl6]·2(HPyCl) (Ce1-1), (HPy)2[CeCl6] (Ce1-2), (HPy)m[Ce38O56−x(OH)xCl50(H2O)12]·nH2O (Ce38), and (HPy)m[Ce52O80−x(OH)xCl59(H2O)17]·nH2O (Ce52) – were crystallized from acidic aqueous solutions using pyridinium (HPy) counterions. The latter consists of two unique cerium oxide nanoclusters that are built from 52 metal ions and represents the largest chloride capped {CeIII/IVO} and/or {MIVO} (M = Ce, Th, U, Np, Pu) nanocluster that adopts the fluorite-type structure of MO2 that has been reported.
Metal oxides are an important class of materials that have found applications ranging from catalysis to biomedicine.1–4 Ceria, CeO2, is one notable example as it is used in catalytic converters, glass polishing, solid-oxide fuel cells, oxygen sensing, alcohol oxidation, biomedical applications, and more.5–8 This wide application space is engendered by the Ce3+/Ce4+ redox-driven formation of highly reactive defect sites composed of oxygen vacancies and Ce3+ ions to form nonstoichiometric CeO2−x.5,8,9 Despite the pervasiveness of ceria-based materials, there is a need to better understand the behavior of bulk ceria for improved reactivity. One way to explore these structure–property relationships is through a molecular lens such as that afforded by cerium–oxo clusters. These species offer well-defined structural models that possess size-dependent properties and also provide insight into the chemical behavior of bulk CeO2.10–12 For example, Estevenon et al. recently demonstrated that Ce–oxo/hydroxo clusters, especially those of higher nuclearities (for example, [Ce38O54(OH)8(CH3CH2CO2)36(C5H5N)8]), could be used as platforms to understand cerium oxide nanomaterials.13 Examination of the structural and electronic properties of Ce clusters and nanoparticles using high-energy-resolution fluorescence detection X-ray absorption spectroscopy (HERFD-XAS), high-energy X-ray scattering (HEXS), and single-crystal X-ray diffraction (SCXRD), showed an evolution in electronic structure as a function of cluster nuclearity, with {Ce38} most closely resembling the properties of CeO2.13 Motivated by these results as well as recent developments in the synthesis and structural chemistry of Ce–oxo clusters, broadly,4,10,12–14 we sought to expand Ce–oxo cluster chemistry by leveraging outer coordination sphere interactions. We have isolated two novel cerium nanoclusters: (HPy)m[Ce38O56−x(OH)xCl50(H2O)12]·nH2O (Ce38) and (HPy)m[Ce52O80−x(OH)xCl59(H2O)17]·nH2O (Ce52) from acidic chloride solutions with pyridinium (HPy1+) counterions. In addition, small changes to the reaction conditions in the presence of HPy1+ allowed for the isolation of monomeric species, (HPy)2[CeCl6]·2(HPyCl) (Ce1-1) and (HPy)2[CeCl6] (Ce1-2). Single crystal X-ray diffraction (SCXRD) was used to elucidate the structural chemistry of the compounds, and Raman and UV-vis-NIR spectroscopies were used to further characterize the isolated phases.
Dissolution of ceric ammonium nitrate in water followed by the addition of ammonium hydroxide resulted in precipitation of cerium hydroxide. The pellet was washed several times with water to remove ammonium. The yellow solid was then dissolved in dilute hydrochloric acid and utilized as a cerium source. Aliquots of pyridine were subsequently added to the Ce solution. Dark yellow crystals of Ce1-1 and Ce1-2, yellow blocks of Ce38, and yellow parallelograms of Ce52 were isolated at room temperature via solvent evaporation. The general synthetic approach follows that described for a {Ce38} nanocluster previously reported by our group,14 but employs pyridinium counterions instead of potassium. Notably, the reaction reproducibly yielded four phases, Ce1-1, Ce1-2, Ce38, and Ce52, in various yields, with the reaction outcome strongly dependent on ambient conditions. In a typical synthesis, anywhere between 5–20 crystals of Ce52 precipitated. Due to the limited yields and co-precipitation of several phases, single crystals, rather than the bulk reaction product, were used for subsequent analyses. Full synthetic details are provided in the ESI.†
Single crystal X-ray diffraction studies revealed the formation of four different phases. Ce1-1, (HPy)2[CeCl6]·2(HPyCl), and Ce1-2, (HPy)2[CeCl6], consist of mononuclear CeCl62− anionic units. In both Ce1-1 and Ce1-2, anionic CeCl62− complexes are charged balanced by two HPy1+ ions in the outer coordination sphere, with Ce1-1 containing two additional HPy1+and two Cl1− ions in the second sphere. Ce1-1 and Ce1-2 were observed when the reaction solutions went to complete dryness. If the solution was not fully evaporated, Ce38 and Ce52 were observed. Ce38 is related to previously reported chloride-capped {Ce38}- and {An38}–oxo clusters (An = U, Np, Pu).14–16 It consists of a cluster core containing 38 Ce sites that is surface decorated by chloride ions and water molecules. Full structure descriptions of Ce1-1, Ce1-2, and Ce38 are provided in the ESI.†
By comparison, Ce52 is built from two discrete cerium-oxo nanoclusters, each containing 52 cerium atoms (Fig. 1). The compound crystallized in the monoclinic space group, P2/m, with the general formula (HPy)m[Ce52O80−x(OH)xCl59(H2O)17]·nH2O. However, two crystallographically distinct {Ce52} clusters, [Ce52O80−x(OH)xCl60(H2O)16]m− and [Ce52O80−x(OH)xCl58(H2O)18]m−, constitute the structure; as reflected in the formula, the clusters differ in the number of chloride and water molecules bound to the surface, and potentially the CeIII/CeIV ratio. For simplicity, only one of the crystallographically unique clusters, [Ce52O80−x(OH)xCl60(H2O)16]m−, is described in detail. The cluster core contains 52 cerium atoms bridged by μ3- and μ4-oxo anions layered in an A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B:C:B:A (A = 6, B = 12, C = 16; based on Ce) pattern (Fig. S8, ESI†), with chloride and water ligands terminating the cluster surface. Ce52 can be broken down into three structural units as shown in Fig. 1. Twenty-four Ce adopt the fluorite-type core of CeO2. Within this {Ce24} subunit, ten of the Ce sites are eight coordinate, CeO8, with the Ce bound exclusively to μ3- and μ4-oxo anions. The remaining fourteen Ce atoms are likewise eight-coordinate; however, these Ce are bound to either one or two water molecules along with oxo anions. Two {Ce6} and four {Ce4} subunits, shown in Fig. 1, complete the Ce52 cluster. Ce atoms in the {Ce6} and {Ce4} units are coordinated to both oxo and chloride anions, with each of the Ce atoms bound to four μ3-/μ4-oxo groups and four chlorides. The chlorides exhibit several coordination modes; there are two μ2-, one μ4-, and one terminal chloro group. Notably, the surface is chloride and water terminated. Average Ce-μ3-O, Ce-μ4-O, Ce–OH2O and Ce–Cl bond distances are 2.240(15) Å, 2.335(14) Å, 2.463(18) Å and 2.863(5) Å, respectively. The average Ce–OH2O, Ce-μ3-O, and Ce-μ4-O bond distances are consistent with those observed previously for a chloride-capped {Ce38}.14 However, the average Ce–Cl bond distance is longer than that reported for a chloride-terminated {Ce38}, which exhibited a Ce–Cl range of 2.683(3)–2.713(3) Å.14
:B:C:B:A (A = 6, B = 12, C = 16; based on Ce) pattern (Fig. S8, ESI†), with chloride and water ligands terminating the cluster surface. Ce52 can be broken down into three structural units as shown in Fig. 1. Twenty-four Ce adopt the fluorite-type core of CeO2. Within this {Ce24} subunit, ten of the Ce sites are eight coordinate, CeO8, with the Ce bound exclusively to μ3- and μ4-oxo anions. The remaining fourteen Ce atoms are likewise eight-coordinate; however, these Ce are bound to either one or two water molecules along with oxo anions. Two {Ce6} and four {Ce4} subunits, shown in Fig. 1, complete the Ce52 cluster. Ce atoms in the {Ce6} and {Ce4} units are coordinated to both oxo and chloride anions, with each of the Ce atoms bound to four μ3-/μ4-oxo groups and four chlorides. The chlorides exhibit several coordination modes; there are two μ2-, one μ4-, and one terminal chloro group. Notably, the surface is chloride and water terminated. Average Ce-μ3-O, Ce-μ4-O, Ce–OH2O and Ce–Cl bond distances are 2.240(15) Å, 2.335(14) Å, 2.463(18) Å and 2.863(5) Å, respectively. The average Ce–OH2O, Ce-μ3-O, and Ce-μ4-O bond distances are consistent with those observed previously for a chloride-capped {Ce38}.14 However, the average Ce–Cl bond distance is longer than that reported for a chloride-terminated {Ce38}, which exhibited a Ce–Cl range of 2.683(3)–2.713(3) Å.14
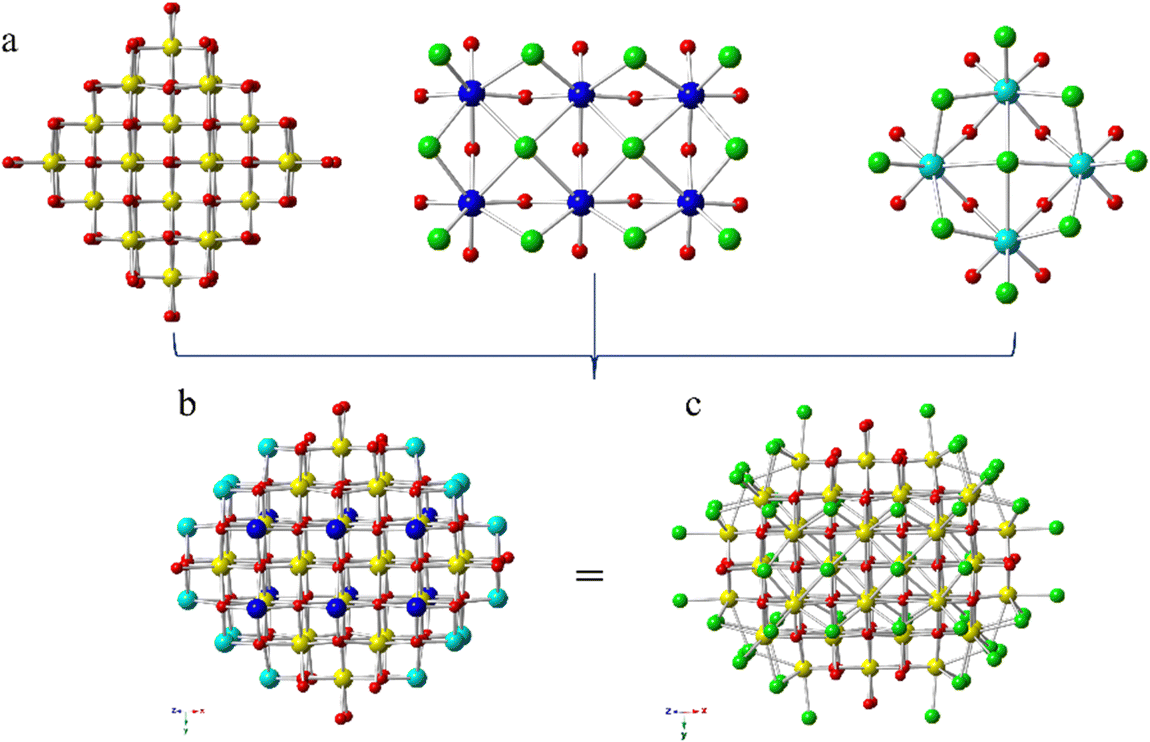 | ||
| Fig. 1 Ball and stick representation of one of the crystallographically unique Ce52 clusters illustrating (a) the {Ce24} (yellow), {Ce6} (dark blue) and {Ce4} (teal) subunits, and (b) the Ce52 core, with the {Ce24}, {Ce6}, and {Ce4} units highlighted. Cl is in green, and O is in red. The chloride/water decorated Ce52 cluster is shown in (c); all of the Ce sites are rendered as yellow spheres. Hydrogen atoms and disorder have been omitted for clarity. | ||
Overall, the {Ce52} cluster is anionic and, as such, protonated pyridinium cations reside in the outer coordination sphere. However, pyridinium ions could not be assigned during the crystal structure refinement due to disorder. Raman spectroscopic data were thus collected on a single crystal of Ce52 (Fig. S13 and Table S12, ESI†). The spectrum was characterized by vibronic stretches indicative of pyridinium, with N–H stretches observed at 1600–1640 cm−1 and C–C stretches at approximately 1000 cm−1. A split peak around 450 cm−1 can be attributed to Ce–O stretching modes; splitting is likely due to the presence of Ce-μ3-O, Ce-μ4-O, and potentially Ce-μ3-OH within the cluster core.14 Notably, for CeO2, Ce–O stretching is typically observed around 465 cm−1, and observation of the peaks around 450 cm−1 as well as a peak at 269.5 cm−1 is consistent with the Raman spectrum for CeO2,17 and underscore the similarities between Ce52 and CeO2. Peaks in the range of 200–400 cm−1 are consistent with Ce–Cl and Ce–OH2O modes.14,18
Bond valence summation (BVS) was used to determine the oxidation state of the Ce atoms in Ce52 (Table S6, ESI†). Several Ce sites exhibited BVS values less than four, which may indicate some contribution from CeIII. Formulation of the cluster as CeIII/CeIV would be consistent with previously reported Ce nanoclusters. However, ambiguity in these assignments arises from uncertainty in the formulation of the μ3-O sites as O2− or OH−, as well as the number of unresolved pyridinium molecules in the outer coordination sphere. It is also worth noting that recent work in polyoxovanadate clusters has shown that BVS values can deviate from assigned valence states due to delocalized electronic structure.19 Additionally, there is some ambiguity in the experimentally determined BVS parameters for Ce as highlighted in the differences in BVS values calculated for both Ce38 and Ce52 using different BVS parameters (Tables S2–S8, ESI†).20–22 Importantly, Ce–O parameters were recently redetermined.21 This has not been done for Ce–Cl and thus, the assignment of the Ce oxidation state in Ce52 is complicated by the fact that Cl ligands decorate the surface of the clusters.
Given the shortcomings of BVS, the oxidation state of the Ce sites in Ce52 was evaluated by single-crystal UV-vis-NIR electronic absorption spectroscopy (Fig. S13, ESI†). The peak at approximately 1200 nm may be attributed to an intervalence charge transfer band and is consistent with CeIII/CeIV in the structure.14 Cerium complexes can exhibit signals in the UV and visible regions due to 4f1 → 5d1 transitions and ligand-to-metal charge transfer (LMCT) that can occur for CeIII and CeIV, respectively. This is due to CeIII being an f1 metal and CeIV being an f0 with no f → f transitions.14,23 The UV-vis-NIR spectrum also displayed a band with a maximum around 400 nm. This band is likely attributed to LMCT based on literature precedence for halide-CeIV transitions.14,23,24
Based on BVS values and the electronic absorption spectrum, Ce52 likely contains both CeIII/CeIV. BVS values suggest that the CeIV sites are located at the center of the cluster and the possible CeIII sites are located at the surface (Table S5, ESI†). Mixed valent Ce–oxo clusters have been previously reported. Examples include {Ce38}, {Ce40}, and {Ce100} clusters.10,12 For {Ce40} and {Ce100}, CeIII were likewise located at the surface sites and specifically, on the (100) facet.10,12 The presence of CeIII at the surface of Ce–oxo clusters is likely attributed to the ease of formation of oxygen vacancies at the surface rather than in the core.10
Further bulk analysis of Ce52 was complicated by low yields, and the isolation of the compound with other phases including Ce38, Ce1-1, and Ce1-2 (Fig. 2); the latter consists of CeCl62− structural units. The isolation of various phases upon minor changes to the reaction conditions suggests that small differences in energy or solubility separate the formation of these compounds. Yet, the factors that push the reaction product to one phase over another remain unclear as solvent evaporation presumably impacts the concentration, pH, and ionic strength of the solution. Interestingly, Ce52 and Ce38 were observed when little solution was left, and Ce1-1 or Ce1-2 were isolated upon complete evaporation of the mother liquor. It is also worth noting that Ce52 crystals left in solution would often redissolve and subsequently precipitate as Ce38.
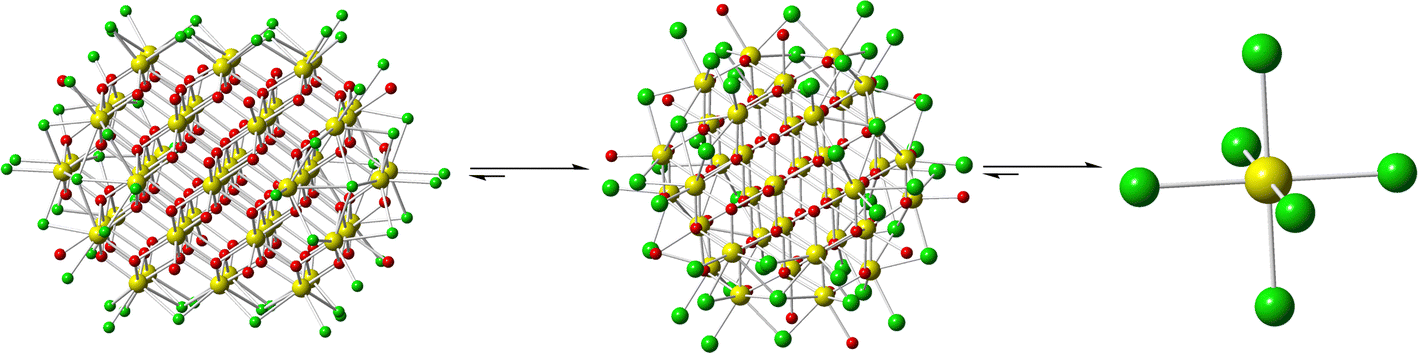 | ||
| Fig. 2 Illustration of the structural units isolated via solvent evaporation: Ce52 (left), Ce38 (middle), and CeCl62− (right). | ||
Several Ce–oxo units ranging from {Ce2} to {Ce100} have been isolated to date, with {Ce6} being the most common cluster core reported for Ce.10–12,14,25–36 These clusters vary in decoration; most are capped by organic ligands (e.g., benzoic acid, propionic acid, and acetic acid), but inorganic anion (e.g., chloride, sulfate) decorated clusters have also been observed.4,10,11,14 Interestingly, clusters with nuclearities larger than {Ce6}, including {Ce24}, {Ce38}, {Ce40} and {Ce100},10,12 typically adopt the cubic, fluorite-type core of bulk CeO2. Indeed, the Ce38 and Ce52 clusters reported here adopt the same fluorite-type structure. As mentioned previously, Ce38 relates to previously reported {Ce38} clusters.10,11,14Ce38 also parallels other {M38} clusters that have been reported for U, Np, and Pu.15,16,37–40 In contrast to Ce38, no other {Ce52} or {An52} clusters that adopt the same core structure as Ce52 described herein have been reported. For transition metal and lanthanide ions, including M = Pd, Au, Ti, Co, Cu, Ag, Eu, Nd, Pr, Gd, and Dy, {M52} clusters have been described.41–48 However, these clusters adopt altogether different topologies than Ce52, and some are heterometallic, such as the Eu52Ni56−xCdx cluster reported by Zheng et al.48 Moreover, although Ce–oxo cluster chemistry literature has grown considerably over the past 10 years, there is a notable gap between {Ce40} and {Ce100}. Furthermore, no inorganic ligated clusters larger than Ce38 that adopt the fluorite-type structure of CeO2 have been isolated. Density Functional Theory calculations have sought to examine the formation energies of single oxygen vacancies in {Ce44} and {Ce85}, but importantly, no experimental data have been reported for these compounds.49 As such, the synthesis and structural characterization of Ce52 fills an important gap in our existing knowledge of Ce–oxo cluster chemistry and provides metrical information that may inform on important aspects of metal oxide and actinide chemistry, broadly.15
In summary, we report the synthesis of the largest known chloride-capped cerium cluster, {Ce52}, along with a crystallographically unique {Ce38}–oxo cluster and two CeCl62− monomers. All of these units are anionic and contain pyridinium cations in the outer coordination sphere. Importantly, the Ce52 cluster fills an important gap in our existing knowledge of Ce cluster chemistry. Isolation of this phase along with Ce38 and CeCl62−via subtle changes to the reaction conditions suggests that small differences in energy, concentration, ionic strength, and/or solubility govern the formation and precipitation of these phases and warrant further examination. Aside from expanding Ce cluster chemistry itself, isolation of Ce52 may also provide important insight (i.e., synthetic parameters and metrical information) with which actinide clusters may be targeted and/or identified.
This work was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Early Career Research Program under Award DE-SC0019190.
Data availability
The data supporting this article including experimental details, crystallographic structure refinements, ORTEP diagrams, structure descriptions, and Raman and UV-vis-NIR spectroscopic data have been included as part of the ESI.† Crystallographic data for Ce1-1, Ce1-2, Ce38, and Ce52 have been deposited at the CCDC under 2359377–2359380† and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Wang, J. Yan, S. Chen and Y. Liu, ChemPlusChem, 2023, 88, e202200462 CrossRef CAS PubMed.
- K. Zhou, G. Ding, C. Zhang, Z. Lv, S. Luo, Y. Zhou, L. Zhou, X. Chen, H. Li and S.-T. Han, J. Mater. Chem. C, 2019, 7, 843–852 RSC.
- D. Van den Eynden, R. Pokratath, J. P. Mathew, E. Goossens, K. De Buysser and J. De Roo, Chem. Sci., 2023, 14, 573–585 RSC.
- I. Colliard, J. C. Brown and M. Nyman, Inorg. Chem., 2023, 62, 1891–1900 CrossRef CAS PubMed.
- J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef.
- M. Flytzani-Stephanopoulos, MRS Bull., 2001, 26, 885–889 CrossRef CAS.
- G. M. Mullen, E. J. Evans, B. C. Siegert, N. R. Miller, B. K. Rosselet, I. Sabzevari, A. Brush, Z. Duan and C. Buddie Mullins, React. Chem. Eng., 2018, 3, 75–85 RSC.
- B. Bhushan, S. Nandhagopal, R. Rajesh Kannan and P. Gopinath, Colloids Surf., B, 2016, 146, 375–386 CrossRef CAS PubMed.
- P. Janoš, J. Ederer, V. Pilařová, J. Henych, J. Tolasz, D. Milde and T. Opletal, Wear, 2016, 362–363, 114–120 CrossRef.
- K. J. Mitchell, K. A. Abboud and G. Christou, Nat. Commun., 2017, 8, 1445 CrossRef PubMed.
- M. C. Wasson, X. Zhang, K.-I. Otake, A. S. Rosen, S. Alayoglu, M. D. Krzyaniak, Z. Chen, L. R. Redfern, L. Robison, F. A. Son, Y. Chen, T. Islamoglu, J. M. Notestein, R. Q. Snurr, M. R. Wasielewski and O. K. Farha, Chem. Mater., 2020, 32, 8522–8529 CrossRef CAS.
- B. Russell-Webster, J. Lopez-Nieto, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2021, 60, 12591–12596 CrossRef CAS PubMed.
- P. Estevenon, L. Amidani, S. Bauters, C. Tamain, M. Bodensteiner, F. Meurer, C. Hennig, G. Vaughan, T. Dumas and K. O. Kvashnina, Chem. Mater., 2023, 35, 1723–1734 CrossRef CAS.
- J. N. Wacker, A. S. Ditter, S. K. Cary, A. V. Murray, J. A. Bertke, G. T. Seidler, S. A. Kozimor and K. E. Knope, Inorg. Chem., 2022, 61, 193–205 CrossRef CAS PubMed.
- L. Soderholm, P. M. Almond, S. Skanthakumar, R. E. Wilson and P. C. Burns, Angew. Chem., Int. Ed., 2008, 47, 298–302 CrossRef CAS PubMed.
- N. P. Martin, C. Volkringer, P. Roussel, J. Marz, C. Hennig, T. Loiseau and A. Ikeda-Ohno, Chem. Commun., 2018, 54, 10060–10063 RSC.
- C. Schilling, A. Hofmann, C. Hess and M. V. Ganduglia-Pirovano, J. Phys. Chem. C, 2017, 121, 20834–20849 CrossRef CAS.
- A. Brandt, Y. U. M. Kiselev and L. I. Martynenko, ZAAC, 2004, 474, 233–240 CrossRef.
- B. E. Petel, W. W. Brennessel and E. M. Matson, J. Am. Chem. Soc., 2018, 140, 8424–8428 CrossRef CAS PubMed.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., 1991, B47, 192–197 CAS.
- P. L. Roulhac and G. J. Palenik, Inorg. Chem., 2003, 42, 118–121 CrossRef CAS PubMed.
- A. Trzesowska, R. Kruszynski and T. J. Bartczak, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 745–753 CrossRef PubMed.
- J. A. Bogart, A. J. Lewis, M. A. Boreen, H. B. Lee, S. A. Medling, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2015, 54, 2830–2837 CrossRef CAS PubMed.
- J. L. Ryan and C. K. Jørgensen, J. Phys. Chem., 1966, 70, 2845–2857 CrossRef CAS.
- X. Wang, K. Brunson, H. Xie, I. Colliard, M. C. Wasson, X. Gong, K. Ma, Y. Wu, F. A. Son, K. B. Idrees, X. Zhang, J. M. Notestein, M. Nyman and O. K. Farha, J. Am. Chem. Soc., 2021, 143, 21056–21065 CrossRef CAS PubMed.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Chem. Commun., 2015, 51, 12578–12581 RSC.
- K. J. Mitchell, J. L. Goodsell, B. Russell-Webster, U. T. Twahir, A. Angerhofer, K. A. Abboud and G. Christou, Inorg. Chem., 2021, 60, 1641–1653 CrossRef CAS PubMed.
- A. Bilyk, J. W. Dunlop, R. O. Fuller, A. K. Hall, J. M. Harrowfield, M. W. Hosseini, G. A. Koutsantonis, I. W. Murray, B. W. Skelton, A. N. Sobolev, R. L. Stamps and A. H. White, Eur. J. Inorg. Chem., 2010, 2127–2152 CrossRef CAS.
- F. Yuan, Z. Gu, L. Li and L. Sha, Polyhedron, 2017, 133, 393–397 CrossRef CAS.
- S. Banerjee, L. Huebner, M. D. Romanelli, G. A. Kumar, R. E. Riman, T. J. Emge and J. G. Brennan, J. Am. Chem. Soc., 2005, 127, 15900–15906 CrossRef CAS PubMed.
- I. L. Malaestean, A. Ellern, S. Baca and P. Kogerler, Chem. Commun., 2012, 48, 1499–1501 RSC.
- S. L. Estes, M. R. Antonio and L. Soderholm, J. Phys. Chem. C, 2016, 120, 5810–5818 CrossRef CAS.
- J. Jacobsen, B. Achenbach, H. Reinsch, S. Smolders, F.-D. Lange, G. Friedrichs, D. De Vos and N. Stock, Dalton Trans., 2019, 48, 8433–8441 RSC.
- R. Das, R. Sarma and J. B. Baruah, Inorg. Chem. Commun., 2010, 13, 793–795 CrossRef CAS.
- L. Mathey, M. Paul, C. Coperet, H. Tsurugi and K. Mashima, Chem. – Eur. J., 2015, 21, 13454–13461 CrossRef CAS PubMed.
- M. P. Coles, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert, Z. Li and A. Protchenko, Dalton Trans., 2010, 39, 6780–6788 RSC.
- N. A. Vanagas, R. F. Higgins, J. N. Wacker, D. R. C. Asuigui, E. Warzecha, S. A. Kozimor, S. L. Stoll, E. J. Schelter, J. A. Bertke and K. E. Knope, Chem. – Eur. J., 2020, 26, 5872–5886 CrossRef CAS PubMed.
- L. Chatelain, R. Faizova, F. Fadaei-Tirani, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2019, 58, 3021–3026 CrossRef CAS PubMed.
- G. E. Sigmon and A. E. Hixon, Chem. – Eur. J., 2019, 25, 2463–2466 CrossRef CAS PubMed.
- C. Falaise, C. Volkringer, J. F. Vigier, A. Beaurain, P. Roussel, P. Rabu and T. Loiseau, J. Am. Chem. Soc., 2013, 135, 15678–15681 CrossRef CAS PubMed.
- A. Eichhöfer and D. Fenske, J. Chem. Soc., Dalton Trans., 1998, 6, 2969–2972 RSC.
- W. H. Fang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 7480–7483 CrossRef CAS PubMed.
- Q. Lin, J. Li, Y. Dong, G. Zhou, Y. Song and Y. Xu, Dalton Trans., 2017, 46, 9745–9749 RSC.
- H. D. Mai, P. Kang, J. K. Kim and H. Yoo, Sci. Rep., 2017, 7, 43448 CrossRef PubMed.
- S. Zhuang, L. Liao, M.-B. Li, C. Yao, Y. Zhao, H. Dong, J. Li, H. Deng, L. Li and Z. Wu, Nanoscale, 2017, 9, 14809–14813 RSC.
- X. Zou, Y. Lv, X. Kang, H. Yu, S. Jin and M. Zhu, Inorg. Chem., 2021, 60, 14803–14809 CrossRef CAS PubMed.
- E. G. Mednikov, S. A. Ivanov, I. V. Slovokhotova and L. F. Dahl, Angew. Chem., Int. Ed., 2005, 44, 6848–6854 CrossRef CAS PubMed.
- R. Chen, Z. H. Yan, X. J. Kong, L. S. Long and L. S. Zheng, Angew. Chem., Int. Ed., 2018, 57, 16796–16800 CrossRef CAS PubMed.
- C. Loschen, A. Migani, S. T. Bromley, F. Illas and K. M. Neyman, Phys. Chem. Chem. Phys., 2008, 10, 5730–5738 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, ORTEP, UV-vis-NIR and Raman spectra. CCDC 2359377–2359380. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03144j |
| ‡ Current address: Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, 94720, USA. |
| This journal is © The Royal Society of Chemistry 2024 |