
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromoting exchange coupling in (CpiPr5)2Gd2X3 complexes†
Grégoire
David
 *a,
Boris
Le Guennic
a and
Daniel
Reta
*bcd
*a,
Boris
Le Guennic
a and
Daniel
Reta
*bcd
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes)-UMR 6226, F-35000 Rennes, France. E-mail: gregoire.david@univ-rennes1.fr
bFaculty of Chemistry, The University of the Basque Country UPV/EHU, Donostia, 20018, Spain. E-mail: daniel.reta@ehu.eus
cDonostia International Physics Center (DIPC), Donostia, 20018, Spain
dIKERBASQUE, Basque Foundation for Science, Bilbao, 48013, Spain
First published on 25th September 2024
Abstract
Introducing magnetic coupling between lanthanide ions has been shown to yield better-performing single-molecule magnets (SMMs), as exemplified by the Cp2iPr5Ln2I3 family of compounds (CpiPr5: pentaisopropylcyclopentadienyl, Ln: Gd, Tb, or Dy). This unique coupling is mediated through an unpaired electron hosted in a σ-like orbital, that results from the two 5dz2 Ln ions, and understanding these interactions holds the key to continue advancing the rational design of SMMs. Here, we focus on the Cp2iPr5Gd2I3 spin-only system and apply a recently proposed DFT-based decomposition scheme to assess the chemical and structural factors that affect the magnetic coupling. Based on these, we propose synthetically feasible systems with increased coupling.
The magnetic exchange interaction (J) is a key element in the design of materials and molecules with novel magnetic properties, having been imaginatively leveraged to make advances in a range of fields, such as organic magnetism,1 spintronics,2 qubits3 and single-molecule magnets.4 Of particular interest to this work is the latter, whose defining feature is a capacity to retain magnetisation at the molecular level. While numerous strategies have been put forward across the years to design and synthesize these systems,5 the common goal still is to maximise the time a SMM retains its magnetisation, ideally at ever-increasing temperatures. This is sought after because it leads to coercive fields, which provide the SMM with magnetic bistability and the potential to be used as platforms for high-density information storage devices.6 A well-studied strategy over the last decade has been to force an indirect coupling between lanthanide ions through coupling with a radical moiety, resulting in Ln-radical-Ln architectures.4 Recently, huge coercive fields have been achieved by introducing a strong J between two axial lanthanide ions,7,8 opening up a new and exciting avenue towards better-performing SMMs. This strong J has been realised through an exotic coupling between lanthanide ions and singly occupied σ-like orbital, constituted of the 5dz2 orbitals of both ions. This bonding interaction belongs to class III of the Robin–Day mixed-valence classification9 and results in an intermediate situation between a three-coupled-magnetic-centres case and Hund's rule. Given the central role that J has in improving the properties of these systems, it seems appropriate to develop an understanding of what the governing factors are, as well as proposing strategies that afford its control through chemical modifications. To that end, we apply a decomposition scheme,10 that informs of the different contributions that make up J, to the series Cp2iPr5Gd2X3 (Cp = cyclopentadienyl, iPr = isopropyl, Gd = gadolinium, X = chlorine, bromine, iodine). We also investigate how structural variations of the parent Cp2iPr5Gd2I3 compound7 impact J, establishing clear magneto-structural relationships. We focus on the Gd-derivatives because our methodology is well-defined for spin-only systems, whilst given the separate origins of Ln anisotropy and the interaction between their spin component with the unpaired electron hosted by 5dz2 orbital, we hypothesise that our findings are also of relevance across the whole series.
The present series of compounds implies three magnetic centres (labelled Gd1, Gd2 and σ) and their coupling is described by the Heisenberg–Dirac–van Vleck (HDvV) Hamiltonian ĤHDvV = −2JGd1-σŜGd1·Ŝσ − 2Jσ-Gd2Ŝσ·ŜGd2 − 2JGd1−Gd2ŜGd1·ŜGd2. Magnetic exchange coupling may be interpreted as the competition between three main physical contributions11 with (i) the direct exchange J0 corresponding to the direct exchange integral between two magnetic centres, (ii) the kinetic exchange ΔJKE translating the relaxation of the magnetic centres in the low spin-states and analogous to Anderson's superexchange mechanism and (iii) the spin polarisation ΔJSP reflecting the different responses of the non-magnetic electrons to the different spin distribution of the spin states. J0 favours a parallel alignment of the spin of the magnetic centres while ΔJKE has the opposite effect. The impact of ΔJSP on the coupling depends on the system but is expected to be negligible in transition metal- or lanthanide-based series of compounds due to the local nature of the magnetic orbitals. Recent developments allow extracting these three contributions in density functional theory (DFT) (methodology and computational details are presented in the ESI†), and this strategy has been successfully applied to rationalise couplings in various situations.12 In systems implying more than two magnetic centres, these three contributions are used to determine the overall coupling as JΣ = J0 + ΔJSP + ΔJKE.10
As a starting point, we first calculate and decompose JΣ for the crystal structure of Cp2iPr5Gd2I37 for both the Gd–Gd (JGd−Gd) and Gd-σ (JGd-σ) interactions (Table S1, ESI†). Focusing on JGd−σ, since JGd−Gd is comparatively negligible (JΣ = −1, J0 = 0, ΔJSP = 0 and ΔJKE = −2 cm−1), we find that the originally reported value of 333 cm−1 is dominated by the direct exchange (J0 = 350 cm−1), with the remaining being a negative contribution from the kinetic exchange part (ΔJKE = −17 cm−1) and the spin polarisation part (ΔJSP = 0 cm−1) playing no role. This pattern can be understood given that the exchange integral informs of the spatial overlap of the interacting magnetic orbitals, which in this case is large. In what follows, we perform a series of structural distortions (Fig. 2) and chemical modifications (Fig. 3) on the parent compound, looking for the conditions that concomitantly result in a large J0, a minimised negative contribution from ΔJKE and a positive one from ΔJSP in order to maximise JΣ.
First, we perform a series of distortions involving the Gd–Gd distance while keeping the relative position of all other groups fixed (Table S2, ESI† and Fig. 2 left). As expected, as the distance decreases, JΣ increases thanks to an increase in J0 (+40 cm−1) which is damped by a much faster increase of the negative, yet smaller, ΔJKE values (−17 to −34 cm−1). Similarly, in increasing the distance between gadolinium centres, JΣ decreases due to a reduction in J0 and ΔJKE values. This analysis shows that while reducing the Gd–Gd distance introduces a rapidly growing, detrimental contribution from ΔJKE, its effect is masked by the exceedingly larger J0, suggesting that chemical modifications aimed at reducing the Gd–Gd distance are an effective way to promote the overall exchange.
Whereas the analysis of linear Gd–Gd distance indicated that a preferred strategy would be to bring the lanthanide ions closer, it also showed that pushing them apart makes ΔJKE go towards ever smaller negative values. If this was combined with changes that did not decrease J0, it would represent a way to increase the overall exchange in these compounds. To that end, we looked at how changing the angle between Gd ions and the centroid of the halogen ions (Fig. 1) affects the exchange interaction and its contributions – we note here that this distortion keeps the relative orientation and distance of the Cp ligands intact with respect to the gadolinium. We first performed a distortion where both Gd ions are moved symmetrically relative to the I3 centroid. Fig. 2 (middle) shows significant variations in both J0 and ΔJKE contributions. However, the values of both contributions increase in magnitude resulting in almost no variation of the total coupling JΣ, rendering this strategy ineffective. The final structural variation focuses on symmetrically bending the angles between the Cp external ligand, its centroid (“o” on Fig. 1) and the GdI3Gd moiety – the Cp ligand pivots around its centroid, and the centroid itself does not move. As shown Fig. 2 (right), JΣ presents a quadratic decrease, which results from the same behaviour of the dominating direct exchange contribution. For the kinetic exchange, the magnitude also decreases but with a more linear pattern. Hence, having the Cp ligands at right angles with the GdI3Gd moiety should be a preferable arrangement to maximise the coupling. It may be worth noting that this variation has been done on a model structure close to the X-ray one, explaining why the relative energies are not equal to zero at 90 degrees.
 | ||
| Fig. 1 Schematic representation of the structural and chemical modifications investigated. Blue (denoted o) and red dots represent the centroids of the CpiPr5 and X3 components, respectively. X refers to halogen atoms (X = I, Cl, F). Green dashed lines represent the distortions applied to the structure. | ||
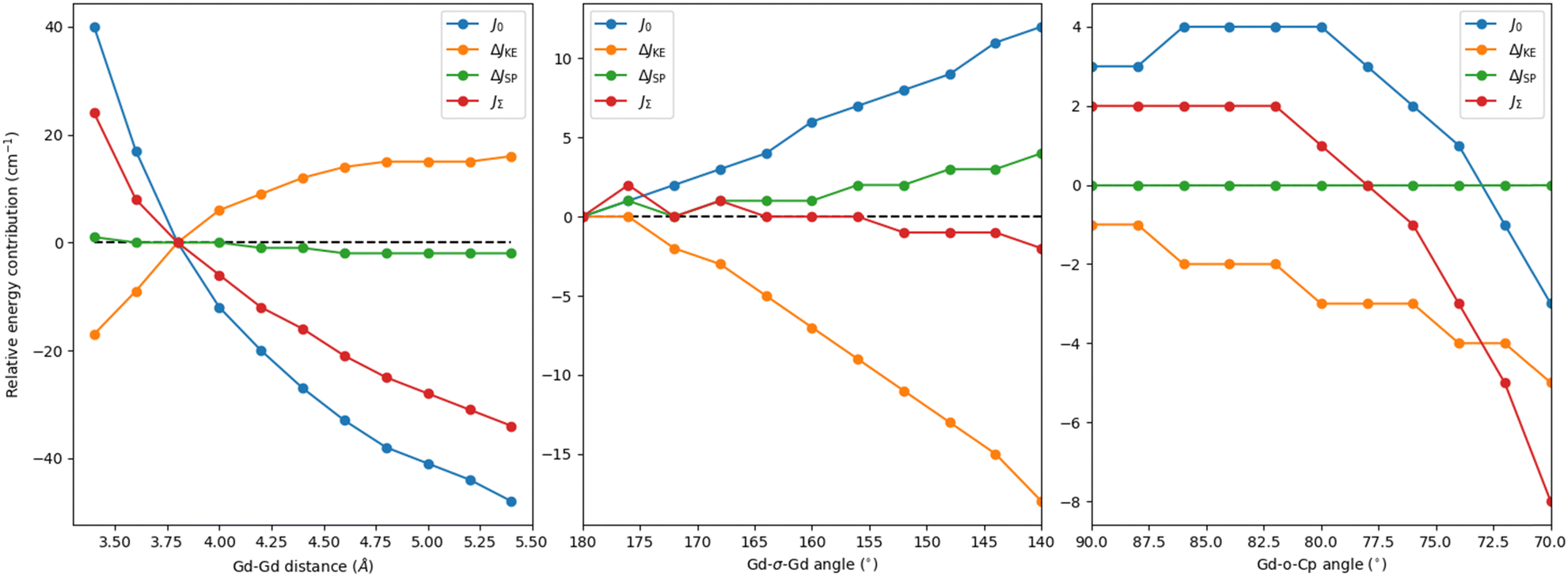 | ||
| Fig. 2 Energy contributions relative to the original Cp2iPr5Gd2I3 complex as a function of the Gd–Gd distance (left), the Gd-σ-Gd angle (middle) and the Gd-o-Cp angle (right) in cm−1. | ||
Despite the strong torsions we applied to the structure, the magnetic exchange coupling between the Gd ions and the σ orbital remains rather unchanged. This may be readily explained by considering the nature of the σ centre; regardless of the torsion applied to the structure, the σ centre would be composed of the 5dz2 orbital of both Gd ions and would result in similar on-site interactions. This peculiarity motivates us to focus on how the bridging halogen triangle affects the σ-like orbital and the associated coupling.
To that end, we first performed the decomposition analysis on the series Cp2iPr5Gd2X3 (X = F, Br, Cl), having simply substituted the original iodine atoms at the crystal structure, without geometry optimisation (Fig. 3 – constrained geometry). Here, we observe an increase of JΣ by substituting with lighter halogen atoms. This trend results from larger direct exchange contributions while the kinetic exchange ones remain rather unchanged. One may also note the larger spin polarisation contribution going from iodine to fluorine. This may be rationalised thanks to the Mulliken spin and charge populations over the gadolinium and halogen atoms presented in Table S3 (ESI†) for X = I and F for the determinants defining the magnetic and core orbitals of the systems (ESI†). In the constrained Cp2iPr5Gd2F3 structure, the spin population is more concentrated over the Gd and less over the halogen than in the Cp2iPr5Gd2I3. One may readily think this would result in a greater on-site interaction, leading to larger J0 and ΔJSP. It may be worth noting that despite this modification of the spin populations, the kinetic exchange contribution is not impacted.
 | ||
| Fig. 3 Contributions to the exchange coupling constants between Gd centre and σ centre in the constrained and relaxed Cp2iPr5Gd2X3 (X = F, Cl, Br, I) structures. | ||
We then performed geometry optimisations on the diamagnetic yttrium analogues and applied our decomposition analysis to those structures. We note that the fluorine substituted system could not be converged to a stationary point. This suggests that the resulting Gd–Gd environment is not capable of effectively hosting a σ-like orbital with the fluorine atoms coming too close to the yttrium during the optimisation. Fig. 3 presents the contributions of the decomposition and JΣ for the series of optimised Cp2iPr5Gd2X3 compounds (relaxed geometry in Fig. 3). Substituting the iodine atoms with lighter halogen atoms results in significantly stronger ferromagnetic J0 contributions, with increases of 39 cm−1 and 56 cm−1 for Br and Cl atoms, respectively. The latter change represents a large modification with an increase of 15%. These chemical substitutions also provide larger ΔJKE in magnitude with lower values of 20 and 24 cm−1 for Br and Cl atoms, respectively. Consequently, these substitutions enhance the ferromagnetic nature of the overall JGd-σ coupling by 23 and 14 cm−1 for Cl and Br, respectively, in line with the Gd–Gd distances (Cl: 3.386, Br: 3.496 Å).
From all the structural distortions and chemical modifications investigated, it is apparent that the best strategy to promote the overall exchange in these compounds is by substituting the heavy iodide ions with smaller halogens, ideally chlorine, as very recently shown in uranium-based triangular complexes.13 In terms of their synthetic feasibility, we argue that the proposed derivatives are reasonable – the original synthesis7 relies on a salt metathesis between anhydrous GdI3 and NaCpiPr5 to form the iodide-bridged dimer precursor, which after reduction, via the formation of potassium iodide (KI), results in the crucial single electron bond between the metals – GdF3,14 GdCl315 and GdBr316 are readily available starting materials, and the reduction of the associated precursor should, in principle, be thermodynamically favoured as the enthalpy of formation of KF, KCl and KBr are 56, 26 and 15 kcal mol−1 higher than that of KI, respectively.
This work focuses on the Gd-based complexes and the study of lanthanide ions with stronger local anisotropy implies tedious theoretical machinery. However, due to the isotropic nature of the magnetic exchange interaction, one may reasonably expect our conclusions to hold for other lanthanide ions and that the increase in J would result in even larger coercive fields and longer relaxation times at temperatures ever closer to ambient conditions. With this, we hope to have provided compelling enough arguments for skilled chemists to take on the challenge of synthesising Dy3Cp2iPr5Cl3 and Tb3Cp2iPr5Cl3.
The authors thank the French GENCI/IDRIS-CINES centres for high-performance computing resources. DR thanks the Basque Government for the IT1584-22 grant. G. D. received research funding from the European Union’s 843 Horizon 2020 Research and Program under Marie Skłdowska-Curie Grant Agreement No. 899546.
Data availability
The data supporting this article have been included as part of the ESI† (geometries and results). Calculations have been performed with a homemade version of ORCA 4, of which the access is restricted, but output files and other data are available upon request from the authors.Conflicts of interest
There are no conflicts to declare.References
- A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503–1505 CrossRef CAS PubMed.
- R. Gaudenzi, E. Burzuri, D. Reta, I. D. P. R. Moreira, S. T. Bromley, C. Rovira, J. Veciana and H. S. J. van der Zant, Nano Lett., 2016, 16(3), 2066 CrossRef CAS PubMed.
- M. Slota, A. Keerthi and W. K. Myers, et al. , Nature, 2018, 557, 691–695 CrossRef CAS PubMed.
- (a) S. Demir, I.-R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef CAS; (b) L. Yang, J. J. Oppenheim and M. Dinca, Dalton Trans., 2022, 51, 8583–8587 RSC; (c) N. Mavragani, A. A. Kitos, J. L. Brusso and M. Murugesu, Chem. – Eur. J., 2021, 27, 5091–5106 CrossRef CAS PubMed; (d) S. Demir, M. I. Gonzalez, L. E. Darago, W. J. Evans and J. R. Long, Nat. Commun., 2017, 8, 2144 CrossRef PubMed; (e) C. A. Gould, E. Mu, V. Vieru, L. E. Darago, K. Chakarawet, M. I. Gonzalez, S. Demir and J. R. Long, J. Am. Chem. Soc., 2020, 142, 21197–21209 CrossRef CAS PubMed; (f) F. Benner, L. La Droitte, O. Cador, B. Le Guennic and S. Demir, Chem. Sci., 2023, 14, 5577–5592 RSC; (g) N. Mavragani, A. A. Kitos, A. Mansikkamäki and M. Murugesu, Inorg. Chem. Front., 2023, 10, 259–266 RSC; (h) S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546–18549 CrossRef CAS PubMed; (i) G. Huang, C. Daiguebonne, G. Calvez, Y. Suffren, O. Guillou, T. Guizouarn., B. Le Guennic, O. Cador and K. Bernot, Inorg. Chem., 2018, 57(17), 11044–11057 CrossRef CAS PubMed; (j) F. Houard, A. Olivier, G. Cucinotta, O. Galangau, M. Gautier, F. Camerel, T. Guizouarn, T. Roisnel, B. Le Guennic, M. Ozerov, Y. Suffren, G. Calvez, C. Daiguebonne, O. Guillou, F. Artzner, M. Mannini and K. Bernot, J. Mater. Chem. C, 2024, 12, 3228–3237 RSC; (k) J. Jung, M. Puget, O. Cador, K. Bernot, C. J. Calzado and B. Le Guennic, Inorg. Chem., 2017, 12, 6788–6801 CrossRef PubMed; (l) J. Rinehart, M. Fang, W. Evans and J. R. Long, Nature, 2011, 3, 538–542 CAS.
- J. L. Liu, Y. C. Chen and M. L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- I. Cimatti, X. Yi, R. Sessoli, M. Puget, B. L. Guennic, J. Jung, T. Guizouarn, A. Magnani, K. Bernot and M. Mannini, Appl. Surf. Sci., 2018, 432, 7–14 CrossRef CAS.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E.-S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS PubMed.
- H. Kwon, K. R. McClain, J. G. C. Kragskow, J. K. Staab, M. Ozerov, K. R. Meihaus, B. G. Harvey, E. S. Choi, N. F. Chilton and J. R. Long, J. Am. Chem. Soc., 2024, 146, 18714–18721 CrossRef CAS PubMed.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 101, 247–422 Search PubMed.
- (a) G. David, G. Duplaix-Rata and B. L. Guennic, Phys. Chem. Chem. Phys., 2024, 24, 8952–8964 RSC; (b) G. David, N. Ferré and B. L. Guennic, J. Chem. Theory Comput., 2023, 19(1), 157–173 CrossRef CAS PubMed; (c) G. David, G. Trinquier and J.-P. Malrieu, J. Chem. Phys., 2020, 153, 194107 CrossRef CAS PubMed.
- J.-P. Malrieu, R. Caballol, C. J. Calzado, C. de Graaf and N. Guihéry, Chem. Rev., 2014, 114(1), 429–492 CrossRef CAS PubMed.
- (a) G. David, F. Wennmohs, F. Neese and N. Ferré, Inorg. Chem., 2018, 57(20), 12769–12776 CrossRef CAS PubMed; (b) G. Duplaix-Rata, B. L. Guennic and G. David, Phys. Chem. Chem. Phys., 2023, 25, 14170–14178 RSC; (c) S. Coetzee, M. M. Turnbull, C. P. Landee, J. C. Monroe, M. Deumal, J. J. Novoa and M. Rademeyer, Phys. Chem. Chem. Phys., 2023, 25, 9176–9187 RSC.
- D. J. Lussier, E. Ito, K. R. McClain, P. W. Smith, H. Kwon, R. Rutkauskaite, B. G. Harvey, D. K. Shuh and J. R. Long, J. Am. Chem. Soc., 2024, 146, 21280–21295 CrossRef PubMed.
- Q. Zhao, W. Lv, N. Guo, Y. C. Jia, W. Z. Lv, B. Q. Shao, M. M. Jiao and H. P. You, Dalton Trans., 2013, 42, 6902 RSC.
- CAS number 10138-52-0.
- CAS number 13818-75-2.
Footnote |
| † Electronic supplementary information (ESI) available: Theory part, computational details, detailed table of data and plot of the magnetic orbitals. See DOI: https://doi.org/10.1039/d4cc03025g |
| This journal is © The Royal Society of Chemistry 2024 |