
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidation of the mechanism of the esterification of boric acid with aliphatic diols: a computational study to help set the record straight†
Corrado
Bacchiocchi
 * and
Manuel
Petroselli‡
*
* and
Manuel
Petroselli‡
*
School of Science and Technology, Chemistry Division, University of Camerino, Via sant'Agostino 1, I-62032, Camerino (MC), Italy. E-mail: corrado.bacchiocchi@unicam.it
First published on 17th October 2024
Abstract
We present a theoretical investigation on the esterification process for boron ester synthesis, considering boric acid and 2(R),4(S)-pentanediol as model boron and diol derivatives, respectively. We report an unprecedented mechanistic pathway, able to rationalise, in contrast with the reported ones, the relatively low Gibbs energies of activation and the pH dependence experimentally observed in the formation of boron esters. We believe that these findings will improve the possibility to predict cross-link reaction rates of boron esters as a fundamental tool in the rational design of functional materials based on boron–oxygen linkages.
The esterification mechanism of boric or boronic acids (boron acids) with diols or polyols is still deemed controversial.1 Computational mechanistic studies reported so far2–10 have not been able to rationalise the dynamic nature of boron-oxygen (B–O) ester linkages formed at room temperature and at neutral pH or under mildly basic conditions (8 < pH < 10). Under these conditions, experimental Gibbs energies of activation range between about 30 kJ mol−1 for aryl boronic acids reacting with aromatic diols,11 and about 70 kJ mol−1 for boric acid reacting with an aliphatic diol,12 with intermediate values of about 50 kJ mol−1 for alkylboronic acids and aliphatic diols.13 Computational studies on these systems, under mildly basic conditions, reported Gibbs energies of activation of about 110 kJ mol−1 or even more,5,7,9,10 highlighting the incompatibility with the well-known dynamic nature of a B–O ester linkage that breaks and reforms relatively quickly at room temperature. Moreover, in some cases, no actual Gibbs energies of activation were reported.6,8
A fundamental understanding of the boron ester dynamic bond formation, similar to those found in some supramolecular systems,14–16 is crucial for the rational design of chemical sensors for saccharide recognition,17 catalytic activators of hydroxy functional groups18 and self-healing hydrogels and polymers based on the B–O linkages.19–22
In all mechanisms proposed so far2–10 (Scheme 1A), each esterification step involves a nucleophilic attack carried out by the oxygen of one hydroxyl group of the diol or polyol (ligand) onto the electron deficient trigonal boron atom, reporting a single transition state (TS). During the B–O bond formation through the single TS, (i) the geometry of the boron atom changes from trigonal to tetrahedral; (ii) a proton transfer occurs from the hydroxyl group of the ligand to the hydroxyl groups of the boron derivative; and (iii) the trigonal geometry of the boron atom is restored after elimination of a water molecule.
 | ||
| Scheme 1 Comparison between the previous (A) and the current proposed mechanism for the esterification process: (B) first esterification step involving the diol and the B(OH)3/B(OH)4− system; (C) OH− transfer between the involved boron derivatives; and (D) second esterification step involving the monoester, previously formed, and the B(OH)4− species. The hydrogen bond interactions are depicted as dashed lines. | ||
Unfortunately, these reported mechanisms did not rationalise the well-known dynamic behaviour in solution at room temperature of B–O ester bonds. Indeed, the esterification rate is reported to be very low under acidic conditions, while it increases by three to four orders of magnitude as the pH becomes greater than the boron acid's pKa.23 This observation prompted us to assume that both the boron acid (trigonal) and the conjugate base (tetrahedral) participate in the rate-determining TS. In light of this, we propose an alternative mechanism for the formation of boron esters (Scheme 1).
To prove our hypothesis, we initially investigated the B(OH)3/B(OH)4− exchange that is known to be relatively fast at room temperature compared to the chemical shift NMR time scale.24,25 A guess geometry for the TS of the B(OH)3/B(OH)4− exchange in water was prepared according to the structure proposed by Ishihara et al.25 assuming a bimolecular mechanism where boric acid and the tetrahydroxyborate anion form two hydrogen bonds in a quasi-symmetric arrangement (Fig. 1). A PES scan of the B–O distance allowed us to determine the TS associated with the initial formation of the bond between the boron atom of boric acid and the oxygen atom of the tetrahydroxyborate's hydroxyl group (Fig. 1, TS1a).
 | ||
| Fig. 1 Structures of the stationary states along the symmetric reaction pathway of the B(OH)3/B(OH)4− exchange at the M062X/aug-cc-pvtz level of theory in water solvent. Atom colours are O: red, H: white, B: pink; chemical bonds forming and breaking are depicted as grey dashed lines, hydrogen bonds as yellow dashed lines, distances are in Å. | ||
By optimising the intrinsic reaction coordinate (IRC) minimum on the product side of TS1a, we found the stationary state I1a and TS1b. Then, a PES scan of the elongation of the B–O bond led us to I1b and, finally, TS1c, associated with the breaking of the B–O bond (Fig. 1). Full computational details can be found in the ESI.† The Gibbs energy of activation of the rate-determining step of the B(OH)3/B(OH)4− exchange, calculated at the CCSD(T)/aug-cc-pvtz//M062X/aug-cc-pvtz level of theory (Fig. S2, ESI†, blue), is 29.1 kJ mol−1. The calculated value is relatively close (7–9 kJ mol−1) to the experimental Gibbs energies of activation for the B(OH)3/B(OH)4− exchange of 20.5 ± 0.8 kJ mol−1,24 and 36.5 kJ mol−1,25 reported in the literature. The value calculated at the M06-2X/aug-cc-pvtz level of theory (Fig. S2, ESI†, black) of 30.7 kJ mol−1 agrees well with both the experimental values and the value obtained at the coupled-cluster level of theory, thus assessing the good accuracy of the M06-2X/aug-cc-pvtz level of theory in the study of these systems. We also found an alternative pathway that proceeds across a single TS (Fig. 2, TS2) where a water molecule mediates the B(OH)3/B(OH)4− exchange. At the same coupled-cluster level of theory, the Gibbs energy of activation of TS2 is 36.9 kJ mol−1, 7.8 kJ mol−1 larger than TS1a, thus adding a small contribution to the exchange rate (see Section 1.3 in the ESI† for details).
 | ||
| Fig. 2 Structure of TS2, an alternative TS of the B(OH)3/B(OH)4− exchange, involving a water molecule, at the M062X/aug-cc-pvtz level of theory in water solvent. Atom colours are O: red, H: white, B: pink; chemical bonds forming and breaking are depicted as grey dashed lines, hydrogen bonds as yellow dashed lines, distances are in Å. | ||
Given the geometry of TS2, we assumed a similar TS geometry for the esterification step where water is substituted by a diol molecule. In practice, we prepared a guess geometry for the first esterification step (Scheme 1B), based on TS2, using boric acid and 2(R),4(S)-pentanediol (2,4-PD), to model a simple 1,3 aliphatic diol. To do this, we positioned a hydroxyl group of 2,4-PD in the place occupied by the water molecule in TS2, obtaining the TS3 structure (Fig. 3). The latter is mainly associated with the proton transfer between one hydroxyl group of the tetrahydroxyborate anion (borate) and one hydroxyl group of the diol. The IRC through TS3 can be described as a “borate-assisted” esterification process between boric acid and one hydroxyl group of the diol. The Gibbs energy of activation of TS3, calculated at the M062X/aug-cc-pvtz level of theory, is 31.5 kJ mol−1, which is the lowest value ever computationally determined for the first esterification step between boric acid and an aliphatic diol. By optimizing the IRC minimum on the product side of TS3 we obtained the stationary state of the tetrahedral borate monoester (Fig. 3, PR3), that was used to explore the intramolecular mechanism related to the formation of a B–O bond in the presence of tetrahedral boron derivatives.
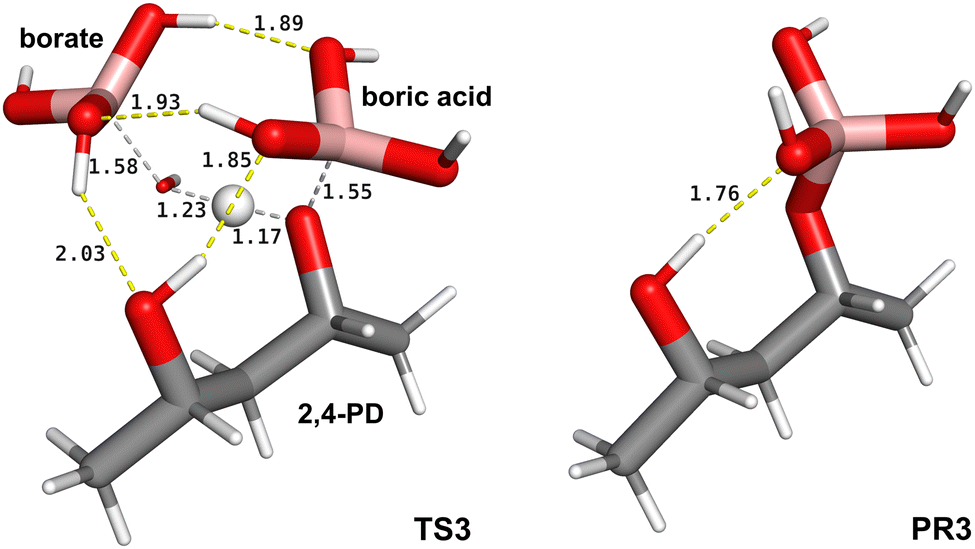 | ||
| Fig. 3 Structure of the TS of the first esterification step between boric acid and 2(R),4(S)-pentanediol (2,4-PD), TS3, and the corresponding tetrahedral borate monoester product, PR3, at the M062X/aug-cc-pvtz level of theory in water solvent. In TS3 the tetrahydroxyborate anion (borate) favours the removal of a proton from one hydroxyl group of the diol. Atom colours are C: grey, O: red, H: white, B: pink; chemical bonds forming and breaking are depicted as grey dashed lines, hydrogen bonds as yellow dashed lines, distances are in Å. | ||
We conducted PES scans of the approach of the tetrahedral boron to the second hydroxyl group of 2,4-PD and, not surprisingly, we did not find any saddle point. This is in agreement with previous computational studies where it is reported that the TSs of the esterification steps always involve trigonal boron.2–10
Therefore, we proceeded to study the tetrahedral/trigonal exchange of the monoester (Scheme 1C) by assuming a reaction pathway similar to that of the B(OH)3/B(OH)4− exchange, determined previously (Fig. 1). As hypothesised, we found an analogous pathway where a boric acid molecule “removes” one of the hydroxyl groups of the tetrahedral monoester, showing a Gibbs energy of activation of 35.6 kJ mol−1, calculated at the M062X/aug-cc-pvtz level of theory. The rate-determining step (TS4a) is shown in Fig. 4 while the remaining stationary states are reported in Fig. S1 (ESI†). The free energy diagram of this pathway (Fig. S4, ESI†) shows that the equilibrium favours the separated reactants (SR4), indicating a stronger acidity of the borate monoester compared to boric acid, in agreement with experimental observations26–31 and computational studies.32 These showed that the pKa differences are predominantly due to electronic effects while a minor contribution is attributed to the trigonal–tetrahedral structure change. The stationary state structure of the trigonal borate monoester is then obtained (Fig. 4, PR4) by optimising the IRC minimum on the product side starting from TS4c (Fig. S1, ESI†). To determine the structure of the second esterification step (Scheme 1D) we decided to take the first step geometry as a template, assuming a similar TS for the formation of the second B–O bond. This led to the TS of the ring closure reaction (Fig. 5, TS5), the corresponding reactant complex (RC5) and the tetrahedral borate cyclic diester product (PR5) being obtained. As observed in TS3, related to the first esterification step, TS5 is mainly associated with the transfer of a proton from the hydroxyl group of the diol to one hydroxyl group of the tetrahydroxyborate anion. The Gibbs energy of activation of TS5 is only 4.7 kJ mol−1, leading to the second esterification process being considered as essentially barrierless. This result is in agreement with early studies on borate esters by Van Duin et al.33 who proposed that the ring closure, in the second esterification step, is relatively faster with respect to the first one. This value is also supported by previous computational mechanistic studies.9
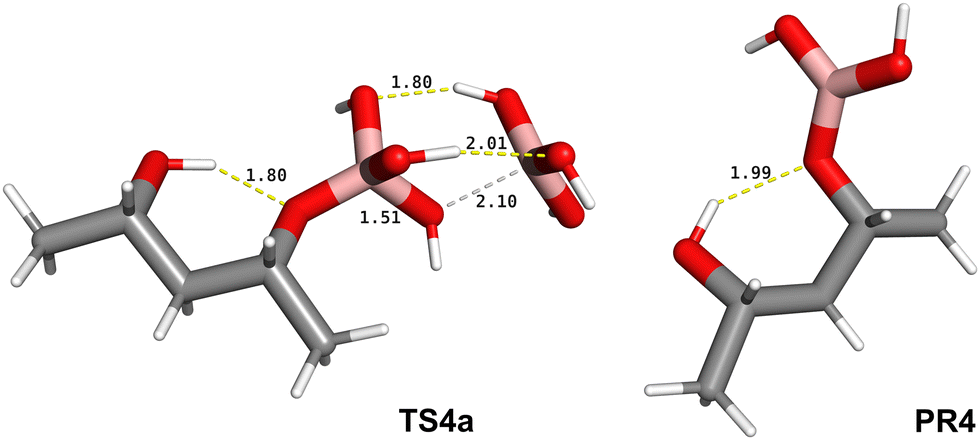 | ||
| Fig. 4 Structure of the rate-determining step of the tetrahedral/trigonal exchange of the borate monoester, TS4a, and of the trigonal borate monoester, PR4, at the M062X/aug-cc-pvtz level of theory in water solvent. Atom colours and bond depictions are as in Fig. 3, distances are in Å. | ||
 | ||
| Fig. 5 Structures of the stationary states along the reaction pathway of the second esterification step between boric acid and 2(R),4(S)-pentanediol, at the M062X/aug-cc-pvtz level of theory in water solvent. The arrangement of the bonds forming and breaking (grey dashed lines) in the TS of this step, TS5, is very similar to that in the first esterification step (Fig. 3, TS3). The reactant complex, RC5, corresponds to the optimised IRC minimum on the reagent side of TS5 and the tetrahedral borate cyclic diester product, PR5, was obtained from the optimised IRC minimum on the product side of TS5. Atom colours and bond depictions are as in Fig. 3, distances are in Å. | ||
In summary, we postulated a reaction pathway, never hypothesised before, for the formation of boron–oxygen (B–O) ester linkages, where both boric acid and tetrahydroxyborate participate in the esterification steps, in contrast to the mechanisms currently reported in the literature,2–10 highlighting the need of a trigonal/tetrahedral boron exchange in a TS separate from those in which the B–O linkages are formed. This mechanism predicts that the rate of formation of the borate diester should exhibit a maximum at a pH close to the pKa of boric acid, where the product of the concentrations of the acid and its conjugate base reaches a maximum. This is in agreement with the seminal work of Yan et al.28 who found that the optimal pH for the binding of a series of boronic acids to diols is close to the pKa of the acid. Our mechanism also predicts a second-order reaction with respect to boron species, promoting more detailed kinetic studies in this direction. To the best of our knowledge, kinetic studies, currently reported on closely related systems,12,13,23,34 did not address this important point. Surprisingly, the proposed mechanism showed that the rate-determining step of the entire esterification process is not the first B–O bond formation, as largely reported,17,30 but the tetrahedral/trigonal exchange of the borate monoester. We believe that the proposed mechanistic study can be applied to the esterification process of a pool of different boron acids and diols. This finding will significantly increase the possibility of predicting the cross-link reaction rates of potential boron esters, and the rational design and fine-tuning of novel self-healing hydrogels and polymers based on the B–O linkages.
C. B.: writing of original draft, writing, data curation, investigation, formal analysis, funding acquisition; M. P.: writing, data curation, formal analysis.
C. B. and M. P. thank professors Marino Petrini, Carlo Santini and Serena Gabrielli (University of Camerino) for useful discussions, MUR for research fundings, and CINFO Computer Centre at the University of Camerino for technical support and assistance.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- S. X. Jin, Y. Q. Li, L. Q. Yang, W. Li and P. Zhou, Anal. Bioanal. Chem., 2023, 415, 2775–2780 CrossRef CAS PubMed.
- K. L. Bhat, S. Hayik and C. W. Bock, J. Mol. Struct.: THEOCHEM, 2003, 638, 107–117 CrossRef CAS.
- K. L. Bhat, S. Hayik, J. N. Corvo, D. M. Marycz and C. W. Bock, J. Mol. Struct.: THEOCHEM, 2004, 673, 145–154 CrossRef CAS.
- K. L. Bhat, V. Braz, E. Laverty and C. W. Bock, J. Mol. Struct.: THEOCHEM, 2004, 712, 9–19 CrossRef CAS.
- H. Monajemi, M. H. Cheah, V. S. Lee, S. M. Zain and W. A. T. W. Abdullah, RSC Adv., 2014, 4, 10505–10513 RSC.
- M. K. Kim, K. H. Eom, J. H. Lim, J. K. Lee, J. D. Lee and Y. S. Won, Korean J. Chem. Eng., 2015, 32, 2330–2334 CrossRef CAS.
- H. F. Li, H. Y. Li, Q. Q. Dai, H. Li and J. L. Brédas, Adv. Theory Simul., 2018, 1, 9 Search PubMed.
- M. Kunimoto, D. Bothe, R. Tamura, T. Oyanagi, Y. Fukunaka, H. Nakai and T. Homma, J. Phys. Chem. C, 2018, 122, 10423–10429 CrossRef CAS.
- V. S. Bharadwaj, M. F. Crowley, M. J. Pena, B. Urbanowicz and M. O'Neill, J. Phys. Chem. B, 2020, 124, 10117–10125 CrossRef CAS PubMed.
- J. Guo, Y. Yang, X. Y. Gao and J. G. Yu, Hydrometallurgy, 2020, 197 Search PubMed.
- B. J. Smith, N. Hwang, A. D. Chavez, J. L. Novotney and W. R. Dichtel, Chem. Commun., 2015, 51, 7532–7535 RSC.
- M. Rietjens and P. A. Steenbergen, Eur. J. Inorg. Chem., 2005, 1162–1174 CrossRef CAS.
- R. Pizer and C. Tihal, Inorg. Chem., 1992, 31, 3243–3247 CrossRef CAS.
- M. Petroselli, S. Mosca, J. Martí-Rujas, D. Comelli and M. Cametti, Eur. J. Org. Chem., 2017, 7190–7194 CrossRef CAS.
- M. Caruso, M. Petroselli and M. Cametti, ChemistrySelect, 2021, 6, 12975–12980 CrossRef CAS.
- M. Petroselli, Y. Q. Chen, M. K. Zhao, J. Rebek and Y. Yu, Chin. Chem. Lett., 2023, 34, 4 CrossRef.
- T. D. James, M. D. Phillips and S. Shinkai, Boronic Acids in Saccharide Recognition, The Royal Society of Chemistry, Cambridge, 2006 Search PubMed.
- D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC.
- Z. H. Zhao, D. P. Wang, J. L. Zuo and C. H. Li, ACS Mater. Lett., 2021, 3, 1328–1338 CrossRef CAS.
- B. Marco-Dufort, R. Iten and M. W. Tibbitt, J. Am. Chem. Soc., 2020, 142, 18730–18731 CrossRef CAS PubMed.
- X. H. Zhao, X. Y. Chen, H. Yuk, S. T. Lin, X. Y. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS PubMed.
- O. R. Cromwell, J. Chung and Z. B. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- R. D. Pizer and C. A. Tihal, Polyhedron, 1996, 15, 3411–3416 CrossRef CAS.
- S. W. Sinton, Macromolecules, 1987, 20, 2430–2441 CrossRef CAS.
- K. Ishihara, A. Nagasawa, K. Umemoto, H. Ito and K. Saito, Inorg. Chem., 1994, 33, 3811–3816 CrossRef CAS.
- D. G. Hall, in Boronic Acids, ed. D. G. Hall, Wiley-VCH Weinheim, 2005, pp. 1–99 DOI:10.1002/3527606548.
- W. A. Marinaro, R. Prankerd, K. Kinnari and V. J. Stella, J. Pharm. Sci., 2015, 104, 1399–1408 CrossRef CAS PubMed.
- J. Yan, G. Springsteen, S. Deeter and B. H. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS.
- G. Springsteen and B. H. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- H. Abu Ali, V. M. Dembitsky and M. Srebnik, Contemporary aspects of boron: chemistry and biological applications, Elsevier B. V, Amsterdam, 2005 Search PubMed.
- M. A. Martínez-Aguirre, F. Medrano, S. Ramírez-Rave and A. K. Yatsimirsky, J. Phys. Org. Chem., 2022, 35, e4425 CrossRef.
- A. Lopalco, V. J. Stella and W. H. Thompson, Eur. J. Pharm. Sci., 2018, 124, 10–16 CrossRef CAS PubMed.
- M. Van Duin, J. A. Peters, A. P. G. Kieboom and H. Van Bekkum, Tetrahedron, 1984, 40, 2901–2911 CrossRef CAS.
- L. Babcock and R. Pizer, Inorg. Chem., 1980, 19, 56–61 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Computational methods, optimised stationary state geometries, energies, energy diagrams, and animations of selected IRC calculations. See DOI: https://doi.org/10.1039/d4cc02999b |
| ‡ Present address: Institute of Chemical Research of Catalonia (ICIQ), Avinguda Països Catalans 16, 43007, Tarragona, Spain. |
| This journal is © The Royal Society of Chemistry 2024 |