
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formal (2+2) ring expansion prevails over (4+2) cycloaddition of a kinetically stabilized benzoborirene with reactive cycloaddends†
Marvin
Sindlinger
a,
Sonja
Biebl
 a,
Markus
Ströbele
b and
Holger F.
Bettinger
*a
a,
Markus
Ströbele
b and
Holger F.
Bettinger
*a
aInstitut für Organische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: holger.bettinger@uni-tuebingen.de
bInstitut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
First published on 23rd August 2024
Abstract
Benzoborirene carrying a bulky Trip2C6H3 (Trip = 2,4,6-tri-iso-Pr3C6H2) group at boron reacts with the dienophile 4-phenyl-1,2,4-triazoline-3,5-dione and the diene 3,6-di(4-pyridyl)-1,2,4,5-tetrazine by opening of the borirene ring rather than undergoing the typical Diels–Alder reactions. The formal insertion results in diazaborole and azaborolo[1,5-b][1,2,4,5]tetrazine derivatives, respectively.
Two fundamental concepts of chemistry, strain and aromaticity, are united in the structural motif of benzoborirene.1 The annulation of the heteroaromatic borirene ring to the aromatic benzene causes high strain and structural deformation of the benzene ring in an anti-Mills-Nixon fashion with a shorter C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C distance between the fusion atoms.1,2 Benzoborirenes only recently became available for investigation as their high reactivity limited access to gas phase and matrix isolation conditions.3–5 While coordination of N-heterocyclic carbenes to the boron atom only provides limited stabilization of A,1 the direct substitution with the electron donating di(trimethylsilyl)amino group of B results in sufficient electronic stabilization for isolation at room temperature (Scheme 1).6
C distance between the fusion atoms.1,2 Benzoborirenes only recently became available for investigation as their high reactivity limited access to gas phase and matrix isolation conditions.3–5 While coordination of N-heterocyclic carbenes to the boron atom only provides limited stabilization of A,1 the direct substitution with the electron donating di(trimethylsilyl)amino group of B results in sufficient electronic stabilization for isolation at room temperature (Scheme 1).6
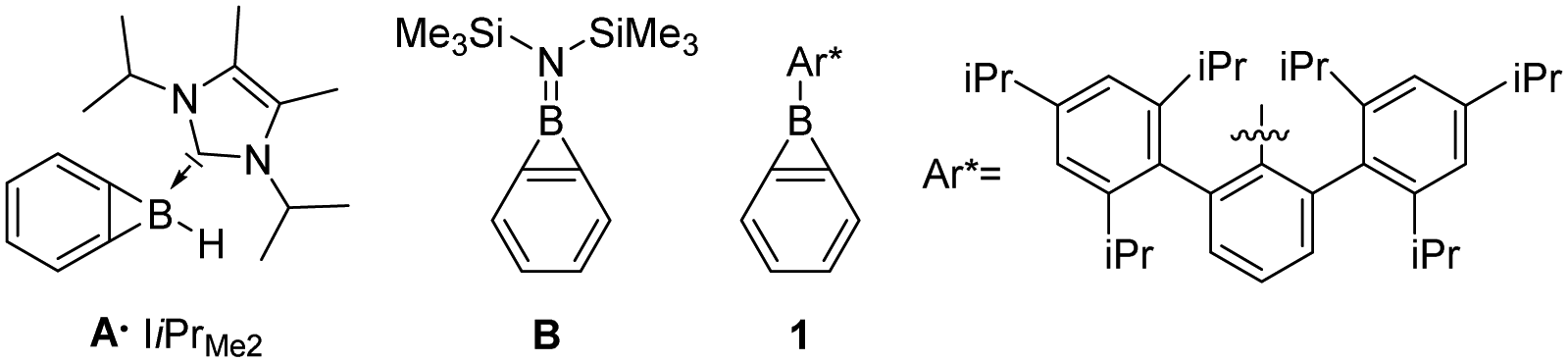 | ||
| Scheme 1 Carbene coordinated benzoborirene A, bis(trimethylsilyl)amino substituted benzoborirene B, and kinetically stabilized benzoborirene 1. | ||
A benzoborirene (1) without substantial electronic stabilization was achieved by employing the sterically demanding Trip2C6H3 (Trip = 2,4,6-tri-iso-Pr3C6H2) substituent.2 The ring system 1 shows high thermal stability, does not react with typical electrophiles such as ketones and trimethylsilyl chloride, and only sluggishly with aldehydes.2,7,8 On the other hand, very rapid reactions are observed for benzoborirenes with the polar multiple bonds of phosphine oxides and isonitriles by formal (2+2) and (2+1) cycloadditions involving a C–B bond.7,8 The related o-carborane-fused aminoboriranes show similar reactivity.8–11
Benzoborirene 1 unites an electrophilic boron atom that experiences steric shielding, a strained three-membered ring, and a slightly distorted benzene ring. In order to investigate the relative reactivity of these units, we focus here on potential cycloaddition reactions of 1 with a highly reactive dienophile and diene, 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) and 3,6-di-(4-pyridyl)-1,2,4,5-tetrazine (4,4′-bptz), respectively. PTAD is among the most reactive bench-stable dienophiles.12,13 It is known to very readily react with cyclopropa[b]naphthalene, an all-carbon analogue of benzoborirene 1, by formal (2+2) cycloaddition to give an indazole derivative presumably involving a zwitterionic intermediate13 (Scheme 2a).14 Triazolinediones are also known to react with strained C–C bonds of, e.g., bicyclo[1.1.0]butane or quadricyclane.15,16
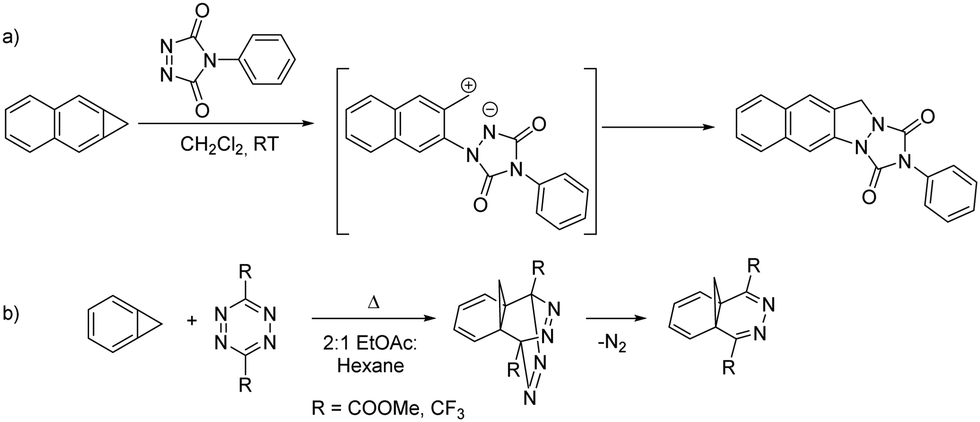 | ||
| Scheme 2 (a) Reaction of cyclopropa[b]naphthalene with PTAD gives an indazol presumably involving a zwitterion according to Halton and Russell;14 (b) reaction of cyclopropabenzene with tetrazines to methanophthalazines as reported by Neidlein and Tadesse.17 | ||
On the other hand, 1,2,4,5-tetrazines carrying electron withdrawing substituents at the 2,6-positions, are very reactive dienes in inverse electron demand Diels–Alder reactions and are thus utilized as bioorthogonal reagents.17–19 Such tetrazines react with cyclopropabenzene to the methano-bridged phthalazine derivative by [π4s + π2s] cycloaddition of the π-bond between the ring fusion atoms, followed by N2 extrusion (Scheme 2b).20
We here report that the behavior of 1 towards PTAD and 4,4′-bptz shows parallels and distinct differences compared to the reactivity reported for cycloproparenes (Scheme 3).
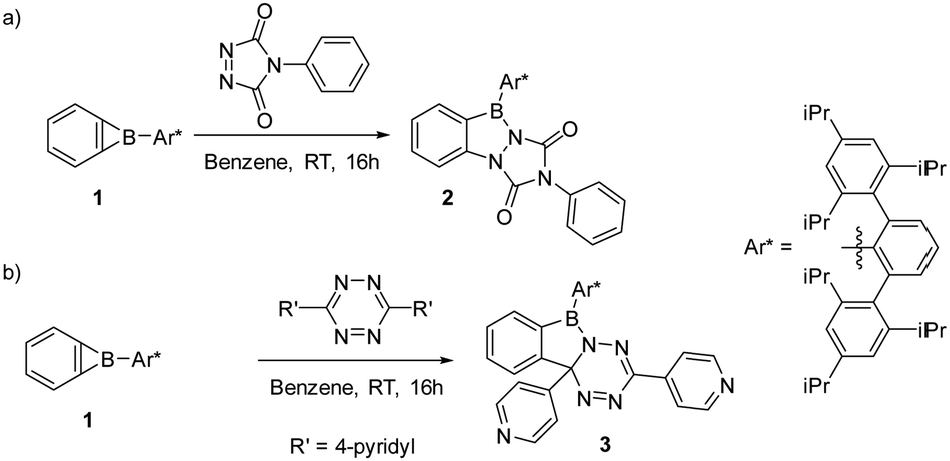 | ||
| Scheme 3 (a) Reaction of 1 with PTAD yields the 2-hydro-1H,5H-benzo[d][1,2,4]triazolo[1,2-a][1,2,3]diazaborole-1,3-dione scaffold 2; (b) reaction of 1 with 3,6-(di-4-pyridyl)-1,2,4,5-tetrazine (4,4′-bptz) yields the 6,10b-dihydrobenzo[3,4][1,2]azaborolo[1,5-b][1,2,4,5]tetrazine derivative 3. | ||
Treatment of 1 with PTAD in benzene over 16 hours results in a pink colored solution. Purification of the crude product by chromatography was not possible on silica, but recycling gel permeation chromatography allowed product isolation in 69% yield. NMR spectroscopy, high-resolution mass spectrometry, and single-crystal X-ray analysis are in agreement with the (2+2) cycloaddition product 2 involving the NN and one C–B bond (Scheme 3a). Single crystals of 2 grew from a saturated pentane solution at −30 °C. Product 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules in the unit cell. The central ring of the terphenyl group is twisted by 91.7° with respect to the planar benzo[d][1,2,3]diazaborol unit (see Fig. 1).
with two molecules in the unit cell. The central ring of the terphenyl group is twisted by 91.7° with respect to the planar benzo[d][1,2,3]diazaborol unit (see Fig. 1).
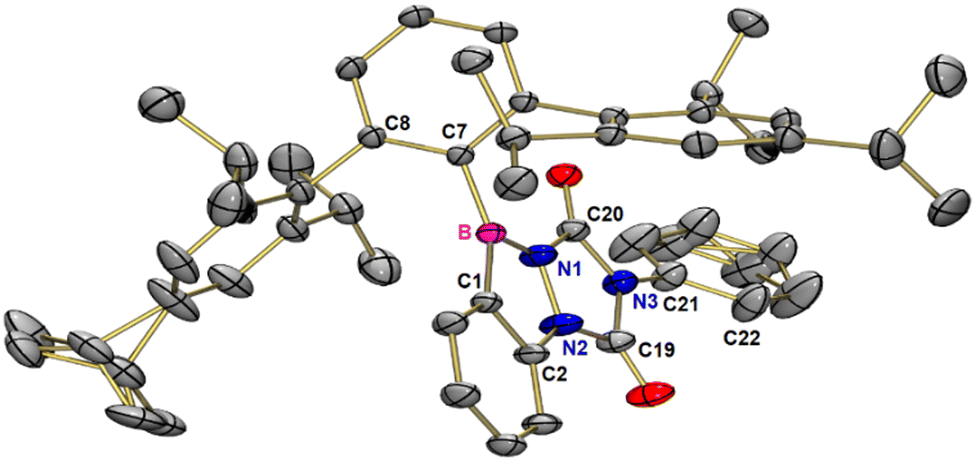 | ||
| Fig. 1 Molecular structure of 2 in the solid state. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths [pm] and bond angles [°] for 2: B–C1 155.4, B–C7 157.0, B–N1 144.8, N1–N2 139.4, N2–C2 138.8, C2–N2–C19–N3 173.84, N1–B–C7–C8 91.74, C19–N3–C21–C22 −41.30. Angle sums [°]: B 360.00, N1 359.81, N2 359.09, C1 359.97, C2 360.00, C19 360.00, C20 360.00, N3 359.86. | ||
The triazole moiety, on the other hand, is slightly kinked (173.8°, ∠C2–N2–C19–N3). The phenyl group is rotated by −41.3° (∠C19–N3–C21–C22) with respect to the triazole unit. All atoms of the five membered rings are in planar trigonal coordination with angle sums of (almost) 360°, with N2 having the smallest angle sum (359.1°). The BN and NN bond lengths are 144.8 ppm and 139.4 ppm, respectively. Their lengths are close to the published values of BN (142.2 pm) and NN (143.7 pm) single bonds in 2,3-dihydro-1,2,3-benzodiazaboroles.21
The reaction of 1 with 3,6-(di-4-pyridyl)-1,2,4,5-tetrazine in benzene at room temperature over 16 hours results in a deep red product mixture. NMR spectroscopy (1H and 13C{1H}) as well as high-resolution mass spectrometry of the crude reaction mixture indicates the presence of one major product 3 (Scheme 3b). We could not obtain product 3 in pure form, as sensitivity to moisture precluded purification by chromatography and preparative scale crystallization was impossible. However, crystals suitable for X-ray structural analysis grew from saturated pentane solution by slow evaporation of the solvent. The main product 3 resulted from formal (2+2) cycloaddition of a C–N bond of the tetrazine to the B–C bond of the benzoborirene ring. Such reactivity is without precedence for 3,6-di-(4-pyridyl)-1,2,4,5-tetrazine.
Product 3 crystallizes in the monoclinic space group Cc with eight molecules in the unit cell and two molecules in the asymmetric unit. In the following, only the (R) enantiomer is considered for a better overview (see Fig. 2). The central ring of the terphenyl unit is rotated by −78.2° (∠N1–B–C7–C8) relative to the benzo[c][1,2]azaborol subunit, while the plane of the tetrazine subunit is rotated by 30.6° (∠C3–C1–N2–N4) relative to the benzo[c][1,2]azaborol plane. The boron atom has trigonal planar coordination with a sum of bond angles of 360.0°. The 4-pyridyl group, bound to the five-membered ring, is rotated by an angle of 125.1° (∠B–C19–C21) relative to the benzo[c][1,2]azaborol plane (see Fig. 2). The N3–N4 bond (126.0 pm) is 10 pm shorter than the N1–N2 bond (136.0 pm). The N2–C20 bond (130.5 pm) is more than 10 pm shorter than the neighboring N4–C20 bond (141.1 pm). This difference is in accord with the Lewis structure drawn in Scheme 3b. For the (S)-enantiomer, the torsion angles are reversed, otherwise, the structural parameters differ only slightly.
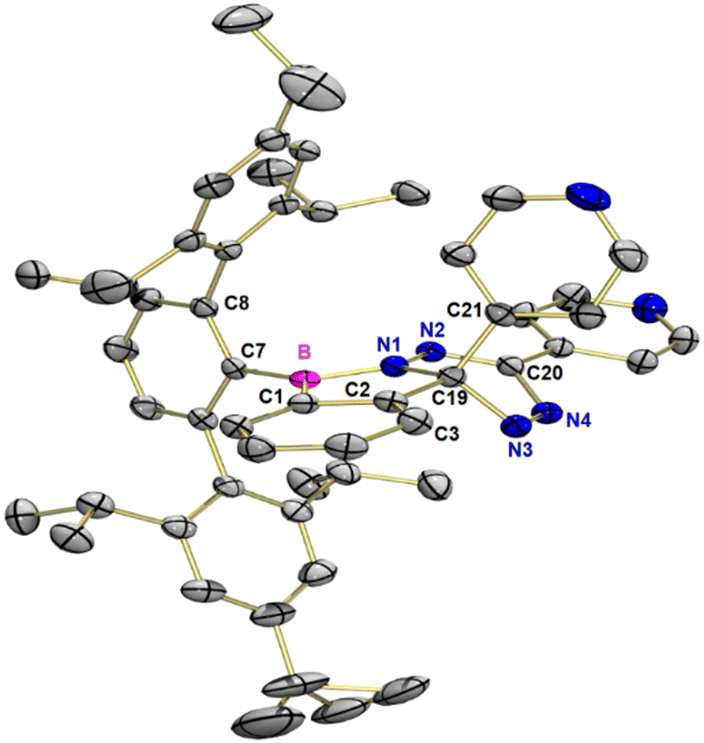 | ||
| Fig. 2 Molecular structure of the (R)-enantiomer of 3 in the solid state. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths [pm] and bond angles [°] for 3: B–C1 155.6, B–C7 157.3, B–N1 144.5, N1–C19 145.4, N3–C19 149.5, C19–C2 151.1, C19–C21 153.2, N1–N2 136.0, N2–C20 130.5, C20–N4 141.1, N3–N4 126.0, B–C19–C21 125.09, N1–B–C7–C8 −78.16, C3–C1–N2–N4 30.60. | ||
Computational chemistry methodology (revDSD-PBEP86-D4/def2-QZVPP/benzene//r2SCAN-3c, see ESI† for details) provides insight into the reactivity of 1 towards PTAD and tetrazine (Fig. 3). Coordination of 1 by a PTAD nitrogen atom is mildly endergonic (ΔG° = 3.0 kcal mol−1) while the transition state TS1 for insertion into the B–C bond is even lower in energy than the complex (ΔG‡ = 2.4 kcal mol−1) indicating a very facile reaction to INT1 (Fig. 3a). The strongly exergonic ring enlargement from INT1 to product 2 (ΔG° = −87.0 kcal mol−1) can proceed over TS2 with a low barrier as well (ΔG‡ = 9.5 kcal mol−1). Hence, the formation of 2 from reagents is a low barrier process that is initiated by B⋯N interaction.
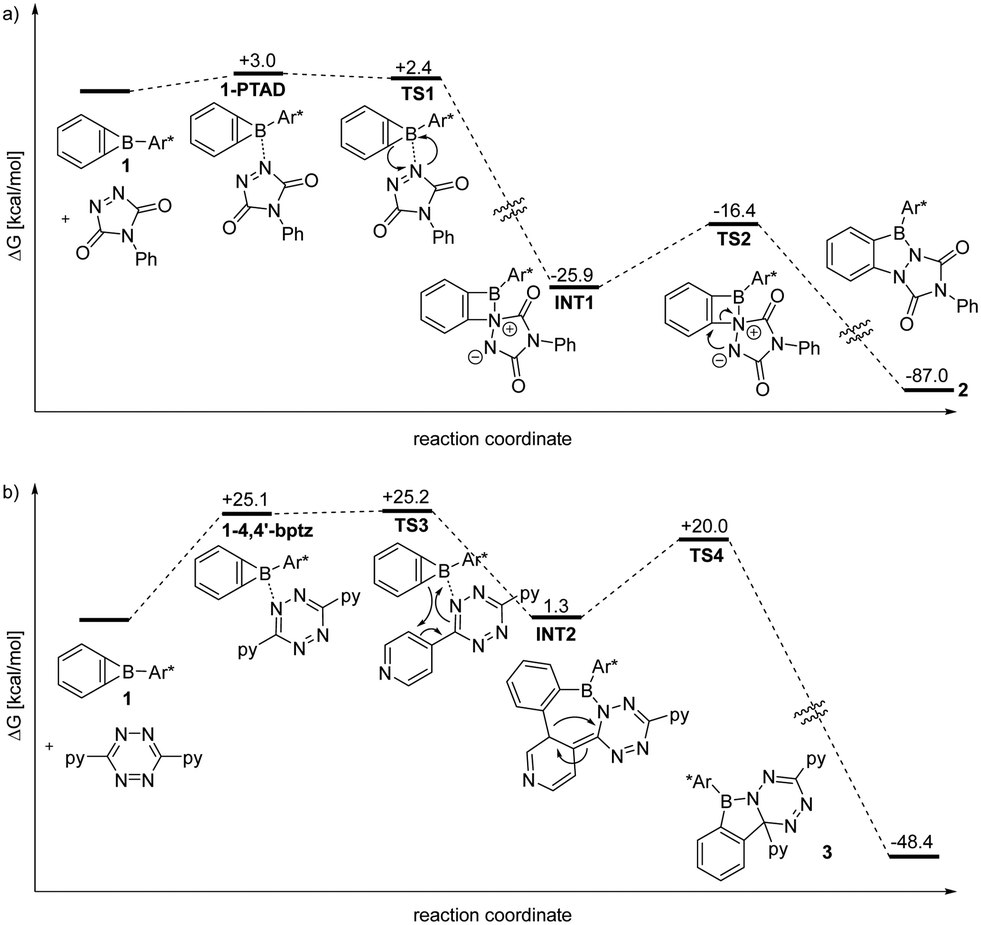 | ||
| Fig. 3 Reaction coordinate (in kcal mol−1) diagrams for the reactions of 1 with PTAD (a) and with 4,4′-bpytz (b) as computed at the revDSD-PBEP86-D4/def2QZVPP/CPCM(DCM)//r2SCAN-3c level of theory. Ar* = Trip2C6H3, py = 4-pyridyl. | ||
A conceivable pathway for the exergonic (ΔG° = −48.4 kcal mol−1) reaction of 4,4′-bptz with 1 to the azaborole 3 that involves an entrance barrier of ΔG‡ = 25.2 kcal mol−1 was identified (Fig. 3b). Coordination of a tetrazine nitrogen atom is endergonic (ΔG° = 25.1 kcal mol−1), but TS3 is very close in energy (ΔG‡ = 25.2 kcal mol−1) and provides INT2 by a formal [4+2] cycloaddition. The strongly contorted INT2 can undergo a phenyl shift (TS4) to give product 3 with a barrier of ΔG‡ = 18.7 kcal mol−1. Although the pyridyl nitrogen atoms are more Lewis basic than those of the tetrazine core, the binding energy of the corresponding adduct (ΔG° = −1.8 kcal mol−1) is sufficiently small to have uncoordinated reagents in solution.
For comparison, we computationally also investigated Diels–Alder reactions between 1 and PTAD and 4,4′-bptz (see ESI,† for details). We find that these reactions involve significantly higher barriers and are mostly strongly endergonic, even when using the phenyl group instead of the large Trip2C6H3 substituent for the modelling.
In summary, we have subjected the kinetically stabilized benzoborirene 1 to reactions with a potent Diels–Alder dienophile (PTAD) and diene (3,6-(4-pyridyl)-1,2,4,5-tetrazine) and observed that in both cases products result from reactions with the borirene unit of 1. The reaction of 1 with PTAD, that parallels the reactivity of cyclopropa[b]naphthalene, provides access to the novel benzo[d][1,2,4]triazolo[1,2-a][1,2,3]diazaborole motif. On the other hand, 1 prefers insertion into the C–B bond with the tetrazine to give an unprecedented benzo[3,4][1,2]azaborolo[1,5-b][1,2,4,5]tetrazine motif rather than following established behavior of cycloproparenes. As both PTAD and 4,4′-bptz are electron poor compounds with low basicity22,23 that preferentially react with electron rich systems, the attack at the sterically shielded boron center is remarkable. Diels–Alder reactions involving the benzene backbone of 1 with the investigated substrates are not competitive due to significantly higher barriers and unfavorable thermodynamics.
M. Sindlinger performed the syntheses, crystallizations, spectroscopy, and initial DFT computations. S. B. performed the purification of 2. M. Ströbele measured and solved the single crystal structures. H. F. B. conceptualized the study, acquired funding, performed DFT computations, and wrote the manuscript.
The authors are very grateful to the German Research Foundation (DFG) for the support of this work (BE 3183/5-3). The computations were performed on the BwForCluster JUSTUS2. The authors acknowledge support from the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG. Sonja Biebl acknowledges support provided by the Fonds der chemischen Industrie for a Kekulé fellowship.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compounds 2 and 3 have been deposited at the Cambridge Crystallographic Data Centre under 2290575 and 2312526.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Hahn, C. Keck, C. Maichle-Mössmer, E. von Grotthuss, P. N. Ruth, A. Paesch, D. Stalke and H. F. Bettinger, Chem. Eur. J., 2018, 24, 18634–18637 CAS.
- M. Sindlinger, M. Ströbele, C. Maichle-Mössmer and H. F. Bettinger, Chem. Commun., 2022, 58, 2818–2821 RSC.
- R. I. Kaiser and H. F. Bettinger, Angew. Chem., Int. Ed., 2002, 41, 2350–2352 CrossRef CAS PubMed.
- H. F. Bettinger, Chem. Commun., 2005, 2756–2757 RSC.
- H. F. Bettinger, J. Am. Chem. Soc., 2006, 128, 2534–2535 CrossRef CAS PubMed.
- H. Zhang, J. Wang, W. Yang, L. Xiang, W. Sun, W. Ming, Y. Li, Z. Lin and Q. Ye, J. Am. Chem. Soc., 2020, 142, 17243–17249 CrossRef CAS PubMed.
- M. Sindlinger, M. Ströbele, J. Grunenberg and H. F. Bettinger, Chem. Sci., 2023, 14, 10478–10487 RSC.
- X. Liu, M. Heinz, J. Wang, L. Tan, M. C. Holthausen and Q. Ye, Angew. Chem., Int. Ed., 2023, 62, e202312608 CrossRef CAS PubMed.
- J. Wang, L. Xiang, X. Liu, A. Matler, Z. Lin and Q. Ye, Chem. Sci., 2024, 15, 4839–4845 RSC.
- J. Wang and Q. Ye, Chem. – Eur. J., 2024, 30, e202303695 CrossRef CAS PubMed.
- Y. Wei, J. Wang, W. Yang, Z. Lin and Q. Ye, Chem. Eur. J., 2023, 29, e202203265 CrossRef CAS PubMed.
- N. Korol, M. Slivka and O. Holovko-Kamoshenkova, Org. Commun., 2020, 13, 146–154 CrossRef CAS.
- K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Chem. Rev., 2016, 116, 3919–3974 CrossRef CAS PubMed.
- B. Halton and S. G. G. Russell, Aust. J. Chem., 1990, 43, 2099–2105 CrossRef CAS.
- M. H. Chang and D. A. Dougherty, J. Org. Chem., 1981, 46, 4092–4093 CrossRef CAS.
- H. Olsen, J. Am. Chem. Soc., 1982, 104, 6836–6838 CrossRef CAS.
- F. Thalhammer, U. Wallfahrer and J. Sauer, Tetrahedron Lett., 1990, 31, 6851–6854 CrossRef CAS.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- R. Neidlein and L. Tadesse, Helv. Chim. Acta, 1988, 71, 249–253 CrossRef CAS.
- M. Shigeno, M. Imamatsu, Y. Kai, M. Kiriyama, S. Ishida, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2021, 23, 8023–8027 CrossRef CAS PubMed.
- J. Spanget-Larsen, J. Chem. Soc., Perkin Trans. 2, 1985, 417–419 RSC.
- W. Kaim and S. Kohlmann, Inorg. Chem., 1987, 26, 68–77 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, spectra, Cartesian coordinates of optimized structures. CCDC 2290575 and 2312526. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02888k |
| This journal is © The Royal Society of Chemistry 2024 |