
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitching between P-acylation and O-acylation of H-phosphonates with chloroformates by changing acyl pyridinium and acyl ammonium ions in a microflow reactor†
Hiroshi
Kitamura
,
Yuma
Tanaka
and
Shinichiro
Fuse
 *
*
Department of Basic Medicinal Sciences, Graduate School of Pharmaceutical Sciences, Nagoya University, Nagoya, 464-8601, Japan. E-mail: fuse.shinichiro.z3@f.mail.nagoya-u.ac.jp
First published on 27th August 2024
Abstract
We report the first switchable acylation of H-phosphonate with chloroformate. The acylation site (P vs. O) in H-phosphonate was switched by changing the acyl pyridinium/ammonium ions. Unexpected phosphite formation was observed during the O-acylation of H-phosphonate. Twenty-six structurally diverse phosphotriesters and phosphonoformate esters were synthesized in microflow reactors.
Organic transformations in which the reaction sites (X and Y) of substrate A can be switched by changing the reaction conditions (Scheme 1a) are useful for synthesizing structurally diverse compounds.1 Although various types of switching of reaction sites have been reported, developing novel and unique switching methods is important because it enables facile access to structurally diverse molecules, leading to the discovery of new functional molecules.
 | ||
| Scheme 1 Concept of site-switchable reactions and acylations of H-phosphonates reported previously and in this manuscript. (a) Switching the reaction site by changing the reaction conditions. (b) P-Acylation and O-acylation of H-phosphonates with acyl chloride or chloroformates (previous reports). (c) Switching between P-acylation and O-acylation of H-phosphonates with chloroformates (this work). | ||
H-Phosphonate E has been used as a building block in the synthesis of organophosphorus compounds.2 Various organic transformations of E have been developed;3 however, the acylation of E remains mostly unexplored. There are six reports for P-acylation of E4a,b and its silylated derivative G4c–f with acyl chlorides F (Scheme 1b(i)), and there are only two reports by Wada et al. that achieved the challenging O-acylation of H-phosphonate equivalent I (Scheme 1b(ii)).5 The acylation using chloroformates K has been even less explored. Only two reports are available on the P-acylation of E and G (Scheme 1b(iii)),6 and no O-acylation reactions have been reported to date (Scheme 1b(iv)). Although modified nucleophiles, such as G and I, have been used in the previously reported acylations to control the reaction sites, no previous acylation approaches have been used to modify electrophiles. Realizing the unachieved switching between the P-acylation and O-acylation of E affords various novel organophosphorus compounds.
We have developed efficient microflow processes7 using highly electrophilic acyl ammonium, acyl pyridinium, and acyl imidazolium ions in the synthesis of peptides and urethane-protected amino acids such as N-carboxy anhydrides.8 A significant change in reactivity has been observed by changing the electrophiles. We also developed the microflow synthesis of acyclic and cyclic phosphotriesters, as well as H-phosphonates.9 Herein, we report the switching of the acylation site (P vs. O) of H-phosphonate E that is in equilibrium with E′ by changing the acyl pyridinium ion N and acyl ammonium ion O in a microflow reactor (Scheme 1c). Various phosphonoformate esters L and phosphotriesters derived from phosphite P were synthesized.
Ethyl chloroformate (1a) and diphenyl H-phosphonate (2a) were employed to examine the switchable acylation of H-phosphonate (Table 1). A microflow reactor was used to precisely control the short reaction times and temperatures of the exothermic processes. Two T-shaped mixers were connected with Teflon® (PTFE: polytetrafluoroethylene; ∅ = 0.80 mm) tubing and were immersed in a water bath. Solutions of 1a in CH2Cl2 and 2a in CH2Cl2 were introduced into the first T-shaped mixer using syringe pumps A and B, respectively. A solution of the nucleophilic amines and i-Pr2NEt was introduced into the second T-shaped mixer using syringe pump C to activate 1a. The resultant mixture was collected in a test tube and stirred for 50 s.10 The reaction was quenched by adding 1 M aqueous HCl.
|
|
||||
|---|---|---|---|---|
| Entry | Nucleophilic amine | 2a (%) | 3a (%) | 5a (%) |
| a Yields were determined by 1H NMR analysis of the crude reaction mixture using 1,1,2-trichloroethane as the internal standard. b THF solution of NMe3 (2 M) was used. NMI = N-methylimidazole; DMAP = 4-dimethylaminopyridine; NMM = N-methylmorpholine; i-Pr = isopropyl; Et = ethyl; Me = methyl; Bn = benzyl. | ||||
| 1 | — | 90 | Trace | n.d. |
| 2 | Pyridine | n.d. | Trace | n.d. |
| 3 | NMI | 36 | 57 | n.d. |
| 4 | DMAP | 19 | 57 | n.d. |
| 5 | 4-Pyrrolidinopyridine | 12 | 55 | n.d. |
| 6 | 9-Azajulolidine | 16 | 74 | n.d. |
| 7 | NMM | 38 | Trace | 45 |
| 8 | Me2NBn | 27 | Trace | 38 |
| 9 | N-Methylpiperidine | 7 | Trace | 83 |
| 10 | N-Methylpyrrolidine | 2 | Trace | 90 |
| 11 | Me2NEt | 3 | Trace | 90 |
| 12 | NMe3b | 1 | Trace | 90 |
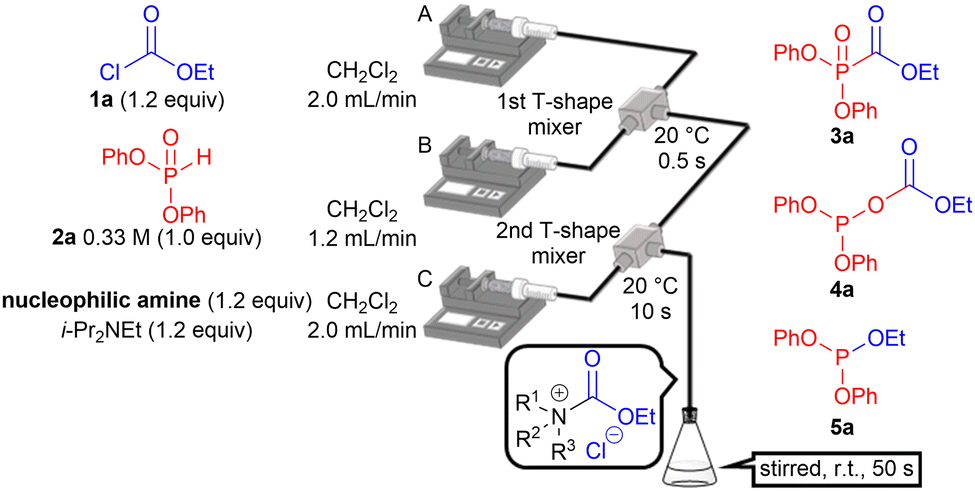
The reaction did not proceed in the absence of a nucleophilic amine (entry 1). Neither P-acylation nor O-acylation occurred, and aminophosphonates were obtained in the presence of pyridine (entry 2). These undesired products were generated via the nucleophilic attack of H-phosphonate on the in situ generated acyl pyridinium ion.11 The use of heterocyclic aromatic amines, including NMI, DMAP, 4-pyrrolidinopyridine, and 9-azajulolidine, afforded 3a (entries 3–6). The highest yield was obtained using 9-azajulolidine (entry 6) and its amount can be reduced to 0.6 equiv (Table S2, ESI,† entry 2) without a decrease in yield. In the presence of aliphatic amines, phosphite 5a was unexpectedly obtained instead of the O-acyl product 4a (entries 7–12). The use of less sterically hindered aliphatic amines including N-methylpyrrolidine, Me2NEt, and NMe3 afforded 5a in higher yields (entries 10–12). We speculated that the in situ generated acyl pyridinium ions or acyl ammonium ions served as electrophiles and their difference in the chemical hardness was the key to the switching of the acylation. The chemical hardness of the electrophiles was estimated using DFT calculations, and the results indicated that the former ions have a lower chemical hardness than the latter ions (for details, see the ESI†). Therefore, it is conceivable that the soft phosphorus atom preferentially reacts with the carbonyl group in the relatively soft acyl pyridinium ions. Reportedly, 9-azajulolidine has a higher electron-donating ability than DMAP.12 We presumed that this delocalizes the positive charge of the acyl pyridinium leading to an increase in the chemical softness. Thus the highest yield was observed. By contrast, the more negatively charged oxygen atom preferentially reacts with localized acyl ammonium ions.
We examined the substrate scope employing optimized conditions.13 In P-acylation (Fig. 1), the electron-withdrawing group on the aromatic ring of H-phosphonate decreased the yield in the synthesis of 3b, whereas the electron-donating group on the aromatic ring of H-phosphonate did not affect the yield in the synthesis of 3c. The desired products were obtained in good yield, regardless of the position of the methyl substituent on the aromatic ring, during the synthesis of 3c–3e. The desired 3f–3i with different sizes of the R′ substituent were obtained in acceptable to good yields. The use of i-Pr2NEt did not result in the acylation of the H-phosphonate 3j containing both alkyloxy and aryloxy groups; therefore, a more basic MTBD was used. Desired 3j was obtained in an acceptable yield. Desired 3k containing two trifluoroethyl groups was also obtained, albeit in a moderate yield. Comparable batch conditions were used to synthesize 3a. (Although a similar yield was observed, gas evolved during the reaction. The batch reaction should be performed with caution. Especially in scale-up synthesis, the use of batch synthesis should be avoided because the reaction is exothermic and is accompanied by the generation of HCl.) The scaled-up synthesis (1.33 g) of 3e was safely performed under flow conditions without a severe decrease in the yield (66%).
 | ||
Fig. 1 Substrate scope for the synthesis of phosphonoformate ester 3. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MTBD was used instead of i-Pr2NEt. The reaction time was extended to 180 s from 120 s. MTBD = 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene. MTBD was used instead of i-Pr2NEt. The reaction time was extended to 180 s from 120 s. MTBD = 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene. | ||
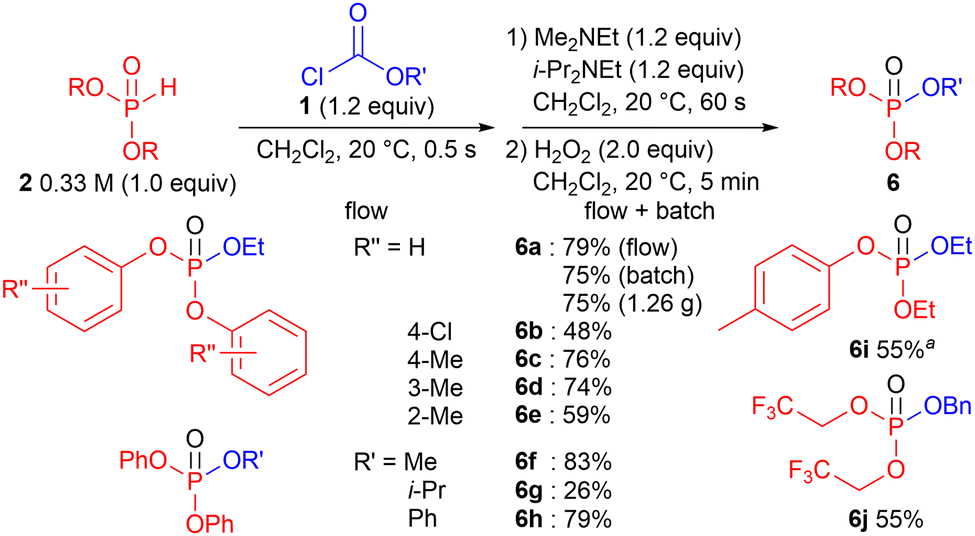
O-Acylation was also examined (Fig. 2).13 The phosphite 5 was oxidized to the corresponding phosphotriester 6 using H2O2 as an oxidant. As in the case of P-acylation, an electron-withdrawing group on the aromatic ring of H-phosphonate decreased the yield of 6b, whereas an electron-donating group on the aromatic ring of H-phosphonate did not affect the yields of 6c and 6d. However, unlike P-acylation, a decrease in the yield was observed in the synthesis of 6e containing two 2-methyl phenoxy groups. The steric bulkiness of OR significantly influenced the product yield. Although 6f with a sterically less hindered methoxy group was obtained in a high yield, 6g with a sterically hindered isopropoxy group was obtained in a low yield, presumably because of the bulky isopropoxy group. Compound 6h with a phenoxy group was obtained in a high yield. The acylation of H-phosphonates containing both alkyloxy and aryloxy groups in the presence of MTBD instead of i-Pr2NEt afforded 6i in a moderate yield. Desired 6j containing two trifluoroethyl groups was also obtained in a moderate yield. Comparable batch conditions were employed for the synthesis of 6a. A slightly lower yield was obtained under batch conditions. (Although a similar yield was observed, the batch reaction should be performed with caution as previously described. Moreover, generation of CO2 is also problematic for O-acylation.) The scaled-up synthesis (1.26 g) of 6a was safely performed under flow conditions without a severe decrease in the yield (75%).
 | ||
| Fig. 2 Substrate scope for the synthesis of phosphotriester 6. aMTBD was used instead of i-Pr2NEt. The reaction time was extended to 240 s from 60 s. | ||
Since commercially available chloroformates are limited, we prepared chloroformate equivalent 9 from triphosgene (7) and alcohol 8 using a modified procedure based on our previous report8g in a microflow reactor, which was used for subsequent O-acylation (Fig. 3). Three T-shaped mixers were connected with Teflon tubing and were immersed in a water bath. A solution of triphosgene (7) in CH2Cl2 and a solution of alcohol 8 and Me2NEt in CH2Cl2 were introduced into the first T-shaped mixer using syringe pumps A and B, respectively. A solution of 2a in CH2Cl2 was introduced into the second mixer using syringe pump C. A solution of Me2NEt and i-Pr2NEt was introduced into the third T-shaped mixer using syringe pump D to activate 9. The resultant mixture was collected in a test tube and stirred for 50 s. The phosphite 5 was oxidized to the corresponding phosphotriester 6 by using H2O2. The reaction was quenched by the addition of 1 M aqueous HCl. The obtained mixtures were purified using column chromatography, preparative TLC, or gel permeation chromatography (GPC).
 | ||
Fig. 3 One-flow synthesis of phosphotriester 6 from alcohol 8via O-acylation. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 1.5 equiv of isopropanol (8a) was used. The reaction time was extended to 120 s from 60 s. 1.5 equiv of isopropanol (8a) was used. The reaction time was extended to 120 s from 60 s. | ||
The desired products 6a and 6k–6n derived from primary alcohol 8 were obtained in good yields. Notably, acid-labile allyl, propargyl, t-butoxycarbonyl, base-labile Fmoc, and alpha-phosphoryl oxy groups were tolerated under the developed conditions. The desired phosphotriester (6g) was obtained in good yield when 1.5 equivalents of the secondary alcohol isopropanol (8a) was added. Comparable batch conditions were used to synthesize 6a. A slightly lower yield was obtained under batch conditions. (The batch reaction should be performed with extreme caution as previously described because the reaction is exothermic and is accompanied by the generation of phosgene, HCl, and CO2.)
We investigated the reaction mechanism for the unexpected formation of phosphite 5a from H-phosphonate 2a and chloroformate 1a by performing several control experiments (for details, see the ESI†). The plausible reaction mechanism for the formation of phosphite P is shown in Scheme 2. The acyl ammonium ion O generated from chloroformate K and aliphatic amine was reacted with tautomerized trivalent H-phosphonate E′ (ref. 14) affording M. Then, intramolecular and intermolecular attacks of the OR′ group concomitant with a decarboxylation of M affords phosphite P. We experimentally confirmed that the externally added ethanol occurred nucleophilic attack instead of the alkoxide (OR′ = OMe) affording P {(RO)2POEt} and this ethanol attack was influenced by the concentration of the reaction mixture (for details, see the ESI†). Thus, we presumed that both intra- and intermolecular attacks of the OR′ group affording phosphite P are possible.
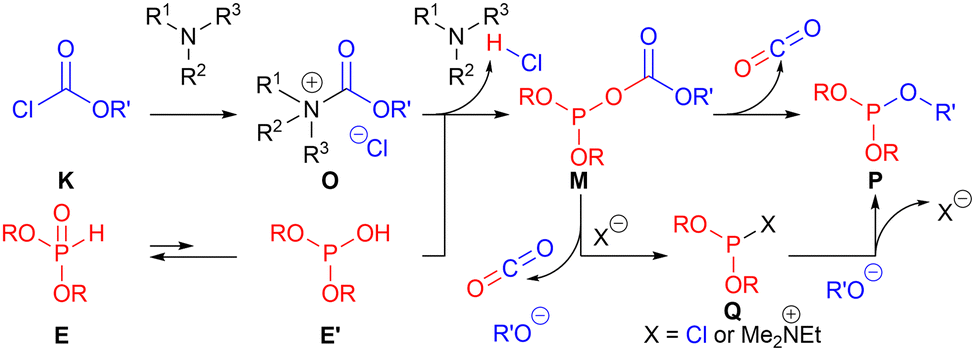 | ||
| Scheme 2 Plausible reaction mechanism for the formation of phosphite. | ||
In summary, we have achieved the first site-switchable acylation of H-phosphonate using chloroformates. In contrast to previously reported approaches, our approach is based on the modification of electrophiles. Acyl pyridinium ions preferentially induced P-acylation, whereas acyl ammonium ions preferentially induced O-acylation. Unexpected and unknown phosphite formation was observed during O-acylation. We suggested that O-acylation and subsequent intra- and intermolecular nucleophilic substitutions afforded phosphite. We also demonstrated the in situ preparation of chloroformates and their use in subsequent O-acylation in a microflow reactor. Twenty-six structurally diverse phosphotriesters and phosphonoformate esters were successfully synthesized. Our developed approach rapidly produced organophosphorus compounds, including unique phosphonoformate esters. This will contribute to library synthesis, leading to the creation of new organophosphorus-based functional materials in the future.
This work was partially supported by the Grant-in-Aid for Scientific Research (B) from JSPS (21H01929), JSPS Fellows (23KJ1102), the Grant-in-Aid for Transformative Research Areas (B) from JSPS (21H05081), JST SPRING (JPMJSP2125), and the Research Support Project for Life Science and Drug Discovery [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from the Japan Agency for Medical Research and Development (AMED) under Grant No. JP23ama121044.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- For recent reviews of switchable reactions, see: (a) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675–1692 RSC; (b) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Soc. Rev., 2020, 49, 7101–7166 RSC ; for recent reports on switchable reactions, see: ; (c) Y. Sakakibara and K. Murakami, ACS Catal., 2022, 12, 1857–1878 CrossRef; (d) C. Ma, J. Shen, C. Qu, T. Shao, S. Cao, Y. Yin, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2023, 145, 20141–20148 CrossRef CAS PubMed; (e) J. Jin, C. Li, R. Wang, Z. Xia, Q. Yan, W. Wang, S. Gu, H. Wang and F. Chen, Org. Lett., 2023, 25, 6012–6017 CrossRef CAS PubMed.
- J. Stawinski and A. Kraszewski, Acc. Chem. Res., 2002, 35, 952–960 CrossRef CAS PubMed.
- A. Kraszewski and J. Stawinski, Pure Appl. Chem., 2007, 79, 2217–2227 CrossRef CAS.
- (a) Y. Ogata and H. Tomioka, J. Org. Chem., 1970, 35, 596–600 CrossRef CAS; (b) T. Yano, T. Kawasaki, T. Yuhki, N. Ishida and M. Murakami, Org. Lett., 2018, 20, 1224–1227 CrossRef CAS PubMed; (c) E. de Vroom, C. E. Dreef, H. van den Elst, G. A. van der Marel and J. H. van Boom, Recl. Trav. Chim. Pays-Bas, 1988, 107, 592–595 CrossRef CAS; (d) T. Johansson and J. Stawinski, Bioorg. Med. Chem., 2001, 9, 2315–2322 CrossRef CAS PubMed; (e) N. Ishida, T. Yano, T. Yuhki and M. Murakami, Chem. – Asian. J., 2017, 12, 1905–1908 CrossRef CAS PubMed; (f) J.-Q. Zhang and L.-B. Han, Org. Lett., 2020, 22, 4633–4637 CrossRef CAS PubMed.
- (a) K. Sato and T. Wada, Org. Biomol. Chem., 2016, 14, 11092–11095 RSC; (b) Y. Takahashi, K. Kakuta, Y. Namioka, A. Igarashi, T. Sakamoto, R. I. Hara, K. Sato and T. Wada, J. Org. Chem., 2023, 88, 10617–10631 CrossRef CAS PubMed . There are two more reports on O-acylation; however, the detailed procedure has not been reported, and acylation is not completely regulated. See: ; (c) A. Kers, I. Kers, J. Stawiński, M. Sobkowski and A. Kraszewski, Tetrahedron, 1996, 52, 9931–9944 CrossRef CAS; (d) A. Kers, I. Kers, A. Kraszewski and J. Stawiński, Collect. Czech. Chem. Commun., 1996, 61, S246–S249 CrossRef CAS.
- (a) R. A. Moss, H. Morales-Rojas, S. Vijayaraghavan and J. Tian, J. Am. Chem. Soc., 2004, 126, 10923–10936 CrossRef CAS PubMed; (b) V. Gagnard, A. Leydet, A. Morère, J.-L. Montero, I. Lefèbvre, G. Gosselin, C. Pannecouque and E. D. Clercq, Bioorg. Med. Chem., 2004, 12, 1393–1402 CrossRef CAS PubMed.
- For selected recent reviews on continuous-flow syntheses, see: (a) R. Gérardy, N. Emmanuel, T. Toupy, V.-E. Kassin, N. N. Tshibalonza, M. Schmitz and J.-C. M. Monbaliu, Eur. J. Org. Chem., 2018, 2301–2351 CrossRef; (b) J. M. De Souza, R. Galaverna, A. A. N. De Souza, T. J. Brocksom, J. C. Pastre, R. O. M. A. De Souza and K. T. De Oliveira, An. Acad. Bras. Cienc., 2018, 90, 1131–1174 CrossRef CAS PubMed; (c) B. T. Ramanjaneyulu, N. K. Vishwakarma, S. Vidyacharan, P. R. Adiyala and D.-P. Kim, Bull. Korean Chem. Soc., 2018, 39, 757–772 CrossRef CAS; (d) P. L. Suryawanshi, S. P. Gumfekar, B. A. Bhanvase, S. H. Sonawane and M. S. Pimplapure, Chem. Eng. Sci., 2018, 189, 431–448 CrossRef CAS; (e) M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC ; for representative publications on “flash chemistry” enabled by flow micro technologies, as reported by Yoshida et al., see: ; (f) J.-I. Yoshida, Flash Chemistry – Fast Organic Synthesis in Micro Systems, Wiley-VCH, Weinheim, 2008 CrossRef; (g) J.-I. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC.
- (a) N. Sugisawa, H. Nakamura and S. Fuse, Chem. Commun., 2020, 56, 4527–4530 RSC; (b) Y. Otake, Y. Shibata, Y. Hayashi, S. Kawauchi, H. Nakamura and S. Fuse, Angew. Chem., Int. Ed., 2020, 59, 12925–12930 CrossRef CAS PubMed; (c) R. Okabe, N. Sugisawa and S. Fuse, Org. Biomol. Chem., 2022, 20, 3303–3310 RSC; (d) O. Shamoto, K. Komuro, N. Sugisawa, T.-H. Chen, H. Nakamura and S. Fuse, Angew. Chem., Int. Ed., 2023, 62, e202300647 CrossRef CAS PubMed; (e) N. Sugisawa, A. Ando and S. Fuse, Chem. Sci., 2023, 14, 6986–6991 RSC; (f) S. Fuse and R. Okabe, Eur. J. Org. Chem., 2023, e202300700 CrossRef CAS; (g) K. Miyamoto, R. Okabe and S. Fuse, Org. Process Res. Dev., 2024, 28, 1971–1978 CrossRef CAS.
- For recent reviews on the microflow synthesis of organophosphorus compounds, see: (a) R. Morodo, P. Bianchi and J.-C. M. Monbaliu, Eur. J. Org. Chem., 2020, 5236–5277 CrossRef CAS; (b) H. Kitamura and S. Fuse, ChemPlusChem, 2023, 88, e202300176 CrossRef CAS PubMed . For our recent reports on the microflow synthesis of organophosphorus compounds, see: ; (c) H. Kitamura, Y. Otake, N. Sugisawa, H. Sugisawa, T. Ida, H. Nakamura and S. Fuse, Chem. – Eur. J., 2022, 28, e202200932 CrossRef CAS; (d) Y. Tanaka, H. Kitamura and S. Fuse, J. Org. Chem., 2024, 89, 1777–1783 CrossRef CAS PubMed; (e) K. Nakabayashi, H. Kitamura and S. Fuse, Chem. – Asian. J., 2024, 19, e202400256 CrossRef.
- The shortening of stirring time in the flask to 10 s significantly decreased the yield (Table S10, ESI,† entry 15). Thus, the desired coupling reaction was not complete in the tube.
- T. Fischer, Q.-N. Duong and O. G. Mancheño, Chem. – Eur. J., 2017, 23, 5983–5987 CrossRef PubMed.
- I. Held, S. Xu and H. Zipse, Synthesis, 2007, 1185–1196 Search PubMed.
- The reaction conditions (reaction times, reaction temperatures, bases, solvents, concentrations, and amounts of reagents) were optimized before the substrate scope. For details, see Tables S2–S4, S6–S10, and S12 (ESI†).
- J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02871f |
| This journal is © The Royal Society of Chemistry 2024 |