
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRed-shifted two-photon-sensitive phenanthridine photocages: synthesis and characterisation†
Célest M.
Attiach
a,
Amit
Kumar
a,
Jonathan
Daniel
b,
Mireille
Blanchard-Desce
 b,
Antoine
Maruani
*a and
Peter I.
Dalko
*a
b,
Antoine
Maruani
*a and
Peter I.
Dalko
*a
aUniversité Paris Cité, CNRS, Laboratoire de Chimie et de Biochimie Pharmacologiques et Toxicologiques, 75006 Paris, France. E-mail: antoine.maruani@cnrs.fr
bUniv. Bordeaux, CNRS, Bordeaux INP, ISM, UMR 5255, F-33400 Talence, France
First published on 15th July 2024
Abstract
Herein we describe the rational design, synthesis and photophysical study of a novel class of phenanthridine-based, one- and two-photon sensitive, photoremovable protecting groups with absorption wavelengths extending beyond 400 nm. This design facilitated the development of scaffolds with enhanced uncaging quantum yield, paving the way for broader applications in controlled drug delivery and molecular manipulation.
Photoremovable protecting groups (PPGs) or photocages represent essential tools for the controlled liberation of biologically active agents through light-mediated covalent bond cleavage.1 Initially prominent in neurophysiology, facilitating the rapid release of neurotransmitters for dynamic signal transmission studies,2 PPGs have evolved to encompass diverse chemical scaffolds.3 From o-nitrobenzyl to BODIPY passing through coumarin-4-ylmethyl,4 these probes have found applications beyond neuroscientific realms, extending to cell physiology3 and optogenetics with photocontrolled release applications for peptides, nucleosides and even proteins5 Recent innovations have further expanded their utility to intricate prodrug approaches and the design of sophisticated drug delivery systems, where light-induced disruption releases cargo in a controlled manner.6
However, challenges persist in the form of limited tissue penetration of light, necessitating solutions like red-shifted absorbing probes and/or the use of two-photon activation at near-infrared wavelengths. This requires the development of specialised probes tailored to these demands.7 Additionally, the field grapples with the intricate balance of criteria governing biological activation, encompassing considerations such as toxicity, water solubility, hydrolytic and metabolic stability, and the efficiency of photolysis. Indeed, the value of the two-photon uncaging cross section, δu, is affected by two parameters: σ2 and ϕu; where σ2 is the two-photon absorption (2PA) cross-section (in GM) and ϕu is the uncaging quantum yield. Increasing σ2 in organic molecules can prove quite challenging as it requires the introduction of multiple large, hydrophobic, aromatic rings that is detrimental to the water solubility of the molecule. On the other hand, ϕu depends on the rate constants of the bond reorganisation events happening after light absorption and can be significantly affected by small structural modifications that have only a slight impact on solubility and that typically do not represent a synthetic challenge.8
In photochemistry, quinolines are among the most responsive PPG under one-photon activation and exhibit interesting sensitivity under two-photon activation conditions.8,9 However, to date, their full potential remains largely underexploited. This is primarily due to their moderate solubility and maximum absorption wavelengths that do not extend beyond 400 nm. By tuning electronic density around quinoline core, it may be possible to improve these characteristics but, despite the synthesis of diverse octupolar10 and quadrupolar probes,11 the increased complexity of such design did not consistently translate into improved performance. Substituents at 7- and 8-positions did not result in major shifts in absorbance but studies on BHQ12 and CyHQ8 by Dore and colleagues revealed acetate cages with uncaging quantum yields up to 0.5.
Interestingly, (dimethylamino)quinoline (DMAQ) and cyclic aliphatic amino derivatives were developed and exhibited even higher uncaging quantum yields but limited maximum absorption wavelengths.13 Recent advancements introduce N-methylquinolinium derivatives as efficient red-shifted PPGs, (i.e. 458 nm) but their low-yielding synthesis and fragmentation rates remains a major limitation.14
In this context, we set out to investigate 3-(dimethylamino)phenanthridine (3-DMAPh) as PPG to provide insights into how “benzannulation” or π-extension influences maximum absorption wavelengths as well as other photophysical properties. To study the impact of electron density modifications, we decided to prepare a small library of 3-DMAPhs derivatives bearing electron-withdrawing (EWG) or electron-donating groups (EDG) (Fig. 1).
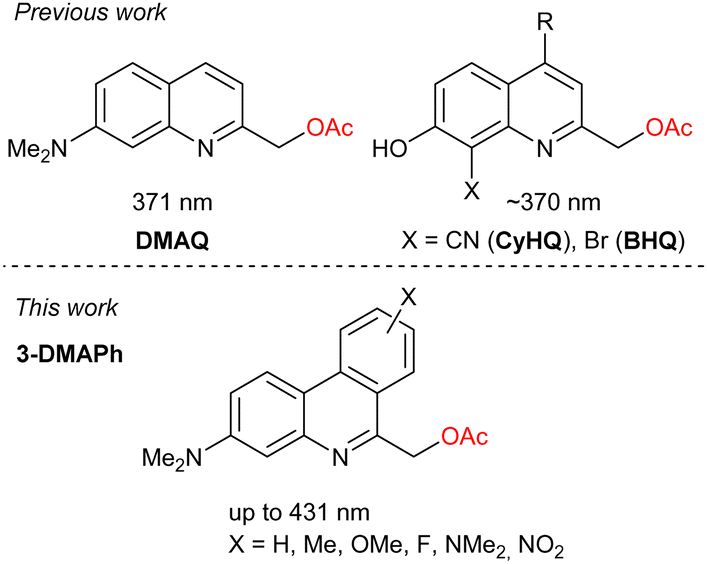 | ||
| Fig. 1 General structure of first and second-generation quinoline-based PPGs (top) and novel phenanthridine-based PPGs (bottom). | ||
These novel PPGs were then used to investigate their photophysical properties and study the parameters regulating their photolytic efficiency. They also served as a basis for developing a DFT-based model to rationalise the development of DMAPh-based PPGs further. Acetate was chosen as a model leaving group for DMAPhs as it is often presented on model substrate and this would enable comparison with existing literature data.8,14,15
The synthesis of 3-DMAPhs started with bromination of acetanilide derivative 116 using (1) eqn of NBS at –50 °C to limit disubstitution. This yielded brominated precursor 2 in 85% yield. Next, a range of commercially available arylboronic acids were coupled through the Suzuki–Miyaura reaction. Optimisation of coupling conditions (see Table S1 in ESI†) enabled the efficient preparation of complete conversion of all arylboronic acids. In addition, while 3a–b necessitated column chromatography, compounds 3c–f could be effectively purified through recrystallisation yielding the products with 54–91% yield.
Subsequently, biphenyls 3a–f were subjected to Bischler-Napieralsky reaction conditions to produce the corresponding phenanthridines (4a–f; Scheme 1). Classical conditions using POCl3 yielded 4a–d with 55–91% yields.17 However, electron-deficient aryls 4e–f exhibited poor cyclisation. A variety of dehydrating agents are available for the Bischler-Napieralsky reaction.18 Among these, Tf2O/Ph3PO (known as the Hendrickson reagent) and a combination of POCl3 and polyphosphoric acid (PPA) were tested (see Table S2 in ESI†).
 | ||
| Scheme 1 Synthesis of 3,6,8 trisubstituted phenanthridine derivatives. Reagents and conditions: (i) Pd(PPh3)4, Na2CO3, THF/H2O, 100 °C, 24 h; (ii) POCl3, reflux, 33 h. | ||
Whilst the Hendrickson reagent proved ineffective, PPA addition significantly improved the cyclisation, resulting in fluoro-phenanthridines (4e) and nitro- (4f) in 83% and 94% yield respectively. Then, oxidation of phenanthridine derivatives 4a–f using selenium oxide gave the corresponding aldehyde in 27–75% yield. With aldehydes 5a–f in hand, reduction/acetylation sequences were carried out (Scheme 2).
 | ||
| Scheme 2 Preparation of –OAc phenanthridine derivatives. Reagents and conditions: (i) SeO2, dioxane, reflux, 3 h; (ii), (iii) NaBH4, Ac2O, 0 °C, 16 h. | ||
In stark contrast with the quinoline-based series8,12,19 it was found that the alcohols obtained from reduction of 6a–f are prone to rapid re-oxidation in ambient air. Despite taking stringent experimental precautions, these efforts proved insufficient to prevent this phenomenon which may have initiated during the work-up process.
To mitigate this unwanted oxidation, it was decided to perform the subsequent acetylation by using NaBH4 both as the reducing agent to prevent reoxidation and as a base in the acetylation step. This allowed to rapidly trap the generated alcohol, resulting in the formation of 6a–e with 38–80% yields.
Interestingly, when reoxidation proved too fast for this sequential approach, performing a one-pot sequence with acetic anhydride as solvent enabled the rapid formation of 6f in 40% yield. Subsequently, compounds 6a–f underwent photochemical characterisation under one- and two-photon excitation (PE).
To assess the impact of substituting 8-substituted phenanthridines-OAc (6a–f), 1PE and 2PE experiments were conducted to assess key photophysical parameters. Phenanthridine 6d, bearing a strong EDG, was not stable in solution and degraded rapidly indicating that a too-high electron-releasing ability is detrimental to dark stability.
In contrast, compound 6f, substituted with a strong EWG, was neither fluorescent nor responsive to irradiation, indicating that a strong intramolecular charge transfer transition is involved, explaining the larger extinction coefficient, low fluorescence quantum yield in a polar solvent like DMSO, as well as the lack of uncaging ability.20 Fortunately, derivatives 6a (H), 6b (Me), 6c (OMe), and 6e (F) could be assessed under both 1PE and 2PE. All compounds exhibited absorption maxima around 400 nm (Table 1 and ESI†), which is encouraging as it represents a 32 nm bathochromic shift compared to 7-DMAQ-OAc. However, their εmax, typically around 2500 M−1 cm−1, was reduced in comparison with 7-DMAQ-OAc.19 Interestingly, once released and as observed during the reduction step of 5a–f, alcohols rapidly reoxidise to aldehydes which were found to display different photophysical properties. Indeed, although 5a–f did not exhibit any fluorescence their maximum absorbance wavelength was significantly red-shifted (Fig. S5, ESI†).
| Compound | λ abs (nm) | λ em (nm) | Stokes shift (×102 cm−1) | ε max (M−1 cm−1) | φ f | λ A2P max (nm) | σ 2 (GM) |
|---|---|---|---|---|---|---|---|
| a Fluorescence quantum yield. Standard: fluorescein in 0.1 M NaOH (φf = 0.9). | |||||||
| 6a (H) | 395 | 544 | 69.3 | 2600 | 25% | 770 | 1.8 |
| 6b (Me) | 395 | 520 | 60.9 | 1900 | 67% | 810 | 1.5 |
| 6c (OMe) | 407 | 528 | 56.3 | 2800 | 30% | 820 | 1.8 |
| 6d (NMe2) | 425 | 520 | 43.0 | 1500 | 44% | — | — |
| 6e (F) | 410 | 547 | 61.1 | 2500 | 24% | 820 | 2.5 |
| 6f (NO2) | 431 | — | — | 5000 | — | — | — |
The presence of the EDG OMe is responsible for slightly higher ε values than the EWG fluorine. EDG and EWG substituents induce bathochromic shifts of the low-energy 1P absorption band compared to reference compound 1b, with increasing shifts with larger ED or EW strengths (Table 1). It should be noted that the UV-vis and fluorescence spectra of compound 6a–e (Table 1) were measured in DMSO whose viscosity mitigates non-radiative decay rate, slows down uncaging and consequently allows to maintain reasonable fluorescence quantum yields. To investigate these experimental results further, computational calculations were conducted.
Indeed, theoretical prediction of the uncaging efficiency presents an attractive approach and offers a way to bypass some experimental cons by providing a relation between chemical/electronic structure and photochemical activity. Although time-dependent density functional theory (TD-DFT) is used extensively to compute nonlinear optical processes for organic molecules due to its reasonable computational cost and relatively good accuracy,21 calculated values may exhibit a complex dependence on other intrinsic molecular parameters, such as transition frequencies and dipole moments, so that the results obtained from different quantum-chemical calculation methods often diverge amongst themselves, as well as deviate from the corresponding experimental values. We therefore benchmarked various functionals and found that M06-2X functional showed good agreement between theoretical and experimental values.20 Thus, DFT and TD-DFT calculations were performed to gain further insight into the polarisation and electronic structures of the ground and first excited states of the investigated series of 3-DMAPhs.
The excitation energies (λ01), one-photon oscillator strengths (f01), dipole moment variation (Δμvert01) and charge transferred upon excitation (qCT) were calculated with TD-DFT but it is important to note that although they are relevant pieces of information, they do not appear to show a direct correlation with uncaging efficiency. However, a correlation can be made between the charge transfer distance (DCT) and uncaging quantum yields.20 The following model was therefore used to calculate and compare these values amongst the molecules of interest (Table 2). Interestingly, extreme values of DCT and Δμvert01 associated with strong EDG (NMe2) and EWG (NO2) did respectively yield to unstable product (DCT = 0.73) and unreactive compound (DCT = 4.74). Whilst the total amount of charge whose distribution is perturbed during electron excitation (qCT) remained relatively stable across the series, in the DCT = [2–3] range, a tendency for improved φu emerges: compounds with lowest DCT values correspond to higher electron-donating strength of substituents. This trend echoes findings observed in coumarin PPG,20 however, the limited diversity of compounds in this study prevents a definitive correlation from being drawn. Nevertheless, it appears more likely that electron-donating groups favour higher values of Φu.
| Compound | λ 01 (λexp) | f 01 | Δμvert01 | q CT | D CT |
|---|---|---|---|---|---|
| a Fudge factor corrected value.22 | |||||
| 6a (H) | 394 (395) | 0.08 | 8.11 | 0.64 | 2.64 |
| 6b (Me) | 396 (400) | 0.09 | 7.48 | 0.63 | 2.49 |
| 6c (OMe) | 407 (407) | 0.09 | 6.53 | 0.61 | 2.23 |
| 6d (NMe2) | 424 (425) | 0.10 | 2.08 | 0.59 | 0.73 |
| 6e (F) | 408 (410) | 0.09 | 8.93 | 0.67 | 2.79 |
| 6f (NO2) | 434 (431) | 0.72 | 18.49 | 0.81 | 4.74 |
Lastly, the 2PA measurements were conducted by registering the two-photon excited fluorescence in DMSO. These spectra revealed the presence of an absorption maximum shifting between 770 nm and 820 nm, depending on the substituent (Table 1). We note that EDG (6c) and EWG (6e) substituents also induce a bathochromic shift of the 2PA band in the NIR, leading to peak 2PA at 820 nm. On the other hand, the substituents do not influence much the 2PA cross-section values, which amount to about 2GM. Though these values remain modest, they are in the same range as those of small DMAQ cages.
To determine the Φu values, irradiation of compounds 6b–c and 6e was performed in 0.1 mM solution in MeCN/Tris 20 mM (1/1, pH 7.4) with LEDs operating at 405 nm. The photochemical reactions were sampled at different time intervals to monitor the progression of the photolysis reaction via HPLC analysis. Overall, t90% ranged from 2 to 10 minutes (Fig. S4, ESI†). Compounds 6a and 6c exhibited similar Φu values (respectively 2.7% and 2.6%) indicating that the methoxy EDG does not affect the uncaging efficiency whereas 6e shows a lower value (0.9%), indicating that EWGs might negatively affect the uncaging quantum yield (Table S3, ESI†). Compound 6b has the largest uncaging efficiency (6%). We note that the EDG did not induce a major bathochromic effect compared to previously described nitrobenzyls.23
The two-photon uncaging cross-section values (δu) fell within a relatively low range in DMSO, in the range of 10−2 GM, and compound 6b exhibited the largest responsiveness, primarily due to its higher quantum yield. To improve this further, DFT calculations were extended to other 3-DMAPhs probes bearing OMe substituents at various positions (Table S4, ESI†) to predict improved photochemical properties. The results suggested that the 7,9-OMe derivative 6g could be the most promising compound among those assayed (DCT = 2.05) To test this hypothesis, it was prepared similarly to the other derivatives (Scheme 1) and its photochemical properties measured (see ESI†). Gratifyingly, as suggested by the DFT calculations, the quantum yield of this dimethoxy derivative reached φu = 10.6%, a 4-times increase compared to unsubstituted phenanthridine 6a. In addition, 6g shows a higher peak 2PA response in the NIR (i.e. 11 GM at 740 nm). Hence the 7,9-OMe substitution pattern leads to both increased uncaging efficiency and 2PA response for DMAPhs, resulting in a two-photon sensitivity of 1.2 GM for compound 6g, 20 times larger than that of 6a (Table S3, ESI†). This suggests that optimal DCT values could be less than but close to 2 to balance δu and stability.
The most efficient compounds proved to be relatively stable towards dark hydrolysis in the photolysis medium, as no significant degradation of the chromophores was observed after several days in solution in the dark at room temperature apart from the one bearing EWG at the 8-position (Fig. S3, ESI†).
In summary, we have developed novel one- and two-photon sensitive PPGs based on a phenanthridine scaffold. The 3-DMAPhs derivatives prepared exhibited a red-shifted absorbance compared to the parent 7-DMAQ derivatives. Both experimental spectroscopic investigation and computational results demonstrated the influence of EDGs and EWGs at various positions on the rings with the best predicted D–π–A–π–D’ candidate showing a 4-time increase in φu and a 5-time increase in σ2max compared to unsubstituted 3-DMAPh. The predictive model along with the straightforward synthesis described here open the route towards rationally-designed red-shifted PPGs based on this novel class of compounds, hence unlocking a variety of applications whilst mitigating the cytotoxic risks of UV radiation.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Bardhan and A. Deiters, Curr. Opin. Struct. Biol., 2019, 57, 164–175 CrossRef CAS PubMed.
- G. C. R. Ellis-Davies, Front. Synaptic Neurosci., 2018, 10, 48 CrossRef CAS PubMed.
- (a) G. C. R. Ellis-Davies, Acc. Chem. Res., 2020, 53, 1593–1604 CrossRef CAS PubMed; (b) G. C. Ellis-Davies, Nat. Methods, 2007, 4, 619–628 CrossRef CAS PubMed.
- (a) P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef CAS PubMed; (b) R. Weinstain, T. Slanina, D. Kand and P. Klan, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- (a) M. Ikeda and M. Kabumoto, Chem. Lett., 2017, 46, 634–640 CrossRef CAS; (b) A. E. Mangubat-Medina and Z. T. Ball, Chem. Soc. Rev., 2021, 50, 10403–10421 RSC.
- W. Zhao, Y. Zhao, Q. Wang, T. Liu, J. Sun and R. Zhang, Small, 2019, 15, e1903060 CrossRef PubMed.
- M. Klausen and M. Blanchard-Desce, J. Photochem. Photobiol., C, 2021, 48, 100423 CrossRef CAS.
- A. K. Hennig, D. Deodato, N. Asad, C. Herbivo and T. M. Dore, J. Org. Chem., 2020, 85, 726–744 CrossRef CAS PubMed.
- B. Kontra, D. Bogdán, B. Alaghehmand, A. Csomos and P. Dunkel, Tetrahedron Lett., 2023, 124 Search PubMed.
- (a) P. Dunkel, M. Petit, H. Dhimane, M. Blanchard-Desce, D. Ogden and P. I. Dalko, ChemistryOpen, 2017, 6, 660–667 CrossRef CAS PubMed; (b) S. Picard, E. Genin, G. Clermont, V. Hugues, O. Mongin and M. Blanchard-Desce, New J. Chem., 2013, 37, 3899 RSC.
- (a) P. Dunkel, C. Tran, T. Gallavardin, H. Dhimane, D. Ogden and P. I. Dalko, Org. Biomol. Chem., 2014, 12, 9899–9908 RSC; (b) C. Tran, N. Berqouch, H. Dhimane, G. Clermont, M. Blanchard-Desce, D. Ogden and P. I. Dalko, Chemistry, 2017, 23, 1860–1868 CrossRef CAS PubMed.
- (a) M. J. Davis, C. H. Kragor, K. G. Reddie, H. C. Wilson, Y. Zhu and T. M. Dore, J. Org. Chem., 2009, 74, 1721–1729 CrossRef CAS PubMed; (b) O. D. Fedoryak and T. M. Dore, Org. Lett., 2002, 4, 3419–3422 CrossRef CAS PubMed.
- B. Kontra, D. Bogdán, B. Alaghehmand, A. Csomos and P. Dunkel, Tetrahedron Lett., 2023, 124, 154587 CrossRef CAS.
- T. Narumi, K. Miyata, A. Nii, K. Sato, N. Mase and T. Furuta, Org. Lett., 2018, 20, 4178–4182 CrossRef CAS PubMed.
- Y. Zhu, C. M. Pavlos, J. P. Toscano and T. M. Dore, J. Am. Chem. Soc., 2006, 128, 4267–4276 CrossRef CAS PubMed.
- H. Kodama, A. Hatanaka, K. Hori and K. Tani, Japan Pat., JP2019038987A, 2019 Search PubMed.
- L. M. Tumir, M. Radic Stojkovic and I. Piantanida, Beilstein J. Org. Chem., 2014, 10, 2930–2954 CrossRef PubMed.
- M. M. Heravi, S. Khaghaninejad and N. Nazari, in Advances in Heterocyclic Chemistry, ed.A. R. Katritzky, 2014, vol. 5, pp. 183–234 Search PubMed.
- M. Petit, C. Tran, T. Roger, T. Gallavardin, H. Dhimane, F. Palma-Cerda, M. Blanchard-Desce, F. C. Acher, D. Ogden and P. I. Dalko, Org. Lett., 2012, 14, 6366–6369 CrossRef CAS PubMed.
- M. Klausen, V. Dubois, G. Clermont, C. Tonnele, F. Castet and M. Blanchard-Desce, Chem. Sci., 2019, 10, 4209–4219 RSC.
- M. A. Salem, I. Twelves and A. Brown, Phys. Chem. Chem. Phys., 2016, 18, 24408–24416 RSC.
- A. Schlachter, A. Fleury, K. Tanner, A. Soldera, B. Habermeyer, R. Guilard and P. D. Harvey, Molecules, 2021, 26, 1780 CrossRef CAS PubMed.
- I. Aujard, C. Benbrahim, M. Gouget, O. Ruel, J. B. Baudin, P. Neveu and L. Jullien, Chemistry, 2006, 12, 6865–6879 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures for the preparation of all products, 1 and 2-photon absorption and fluorescence spectra, details on the photochemical apparatus, characterization data, theoretical data, copies of 1H NMR and 13C NMR spectra for all the synthesized compounds. See DOI: https://doi.org/10.1039/d4cc02852j |
| This journal is © The Royal Society of Chemistry 2024 |