
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClick assembly through selective azaylide formation†
Mayo
Hamada
,
Gaku
Orimoto
and
Suguru
Yoshida
 *
*
Department of Biological Science and Technology, Faculty of Advanced Engineering, Tokyo University of Science, 6-3-1 Niijuku, Katsushika-ku, Tokyo 125-8585, Japan. E-mail: s-yoshida@rs.tus.ac.jp
First published on 5th July 2024
Abstract
An efficient triple-click assembly using a newly designed trivalent platform is disclosed. We achieved the selective azaylide formation of 2,3,5,6-tetrafluorophenyl azides with o-ester-substituted triarylphosphines leaving 2,6-dichlorophenyl azides untouched. Further rapid Staudinger reaction of dichlorophenyl azides and subsequent triazole formation allowed us to prepare trifunctionalized molecules in three steps.
Trivalent platforms having three clickable functional groups for assembling modules gain attention from diverse research fields including pharmaceutical sciences and chemical biology due to the great importance of multifunctional molecules.1–4 Despite recent emerging advances in sequential click reactions, it is not easy to perform click assembly without deprotection due to the limited trivalent platforms. Herein, we disclose an efficient assembly of functional modules using a new trivalent platform through selective robust azaylide formations by Staudinger reaction.
Staudinger reactions have played pivotal roles as efficient conjugation methods for functional molecules.5 Rapid Staudinger reactions forming robust azaylides are gaining attention as efficient click reactions from broad researchers in pharmaceutical sciences and materials chemistry.6–8 Indeed, rapid azaylide formations were developed independently by our group6 and Ramström and Yan's group7 using 2,6-dichlorophenyl azides and 2,3,5,6-tetrafluorophenyl azides, respectively. These rapid reactions allowed us to achieve not only the efficient synthesis of difunctionalized molecules8b,8e but also bioconjugations using living cells due to good biocompatibility.6–8 We conceived that selective azaylide formations of 2,6-dichlorophenyl azides and 2,3,5,6-tetrafluorophenyl azides with phosphines enable us to realize efficient sequential conjugations. In this study, we thus decided to design new platforms having these azide moieties and an alkynyl group based on the combination of azaylide formations and triazole formations9 for the sequential assembly of functional molecules after examining the selective azaylide formations of 2,6-dichlorophenyl azides and 2,3,5,6-tetrafluorophenyl azides with phosphines (Fig. 1).
 | ||
| Fig. 1 (A) Examples of trivalent platforms. (B) Our previous study. (C) Ramström and Yan's work. (D) This work. | ||
We at first performed a competition reaction of an equimolar mixture of 2,6-dichloro-4-(n-butylaminocarbonyl)phenyl azide (1a) and 2,3,5,6-tetrafluoro-4-(n-butylaminocarbonyl)phenyl azide (2a) with triphenylphosphine (3a) in THF at room temperature (Fig. 2). As a result, phosphine 3a was smoothly consumed to afford a mixture of azaylides 4a and 5a quantitatively, where the formation of azaylide 5a from tetrafluorophenyl azide 2a was favored in moderate selectivity. This result obviously demonstrated that tetrafluorophenyl azide 2a showed higher reactivity with triphenylphosphine (3a) than dichlorophenyl azide 1a.
 | ||
| Fig. 2 Competition reaction of azides 1a and 2a with phosphine 3a. | ||
Screening of triarylphosphines 3 resulted in selective azaylide formation using an equimolar mixture of azides 1a and 2a (Table 1). Indeed, we found that treatment of an equimolar mixture of azides 1a and 2a with diphenyl(2-(methoxycarbonyl)phenyl)phosphine (3b) selectively provided azaylide 5 quantitatively from tetrafluorophenyl azide 2a without the reaction of dichlorophenyl azide 1a (entry 1). While electron-donating substituents on the benzene ring of phosphines 3b and 3c decreased the selectivity (entries 2 and 3), electron-withdrawing and slightly bulky substituents such as chloro and bromo group led to the perfect selectivity as well as ester moiety (entries 4–6). When diphenyl(4-(methoxycarbonyl)phenyl)phosphine (3h) was used in the competition reaction, the selectivity was significantly decreased (entry 7). These results clearly showed that the electronic and steric effects of the ester moiety at the ortho position of 3b were key to the success of the selective azaylide formation. Additional substituents such as electron-donating methoxy group and electron-withdrawing ester and amide moieties at the meta position of o-ester-substituted phosphines 3i-3g did not decrease the selectivity (entries 8–10). These results indicated that functions can be installed to the o-(methoxycarbonyl)phenyl group of triarylphosphines. Owing to the rapid Staudinger reaction of 2,3,5,6-tetrafluorophenyl azide with 3b (kobs = 2.7 M−1 s−1),7 this selective azaylide formation will realize broad applications involving bioconjugation.
|
|
|||
|---|---|---|---|
| Entry | 3 | Yield/% (4 + 5) | 4/5 |
| 1 |

|
Quant. | 0/100 |
| 2 |

|
93 | 17/83 |
| 3 |

|
92 | 7/93 |
| 4 |

|
99% | 8/92 |
| 5 |

|
Quant. | 0/100 |
| 6 |

|
Quant. | 0/100 |
| 7 |

|
Quant. | 25/75 |
| 8 |
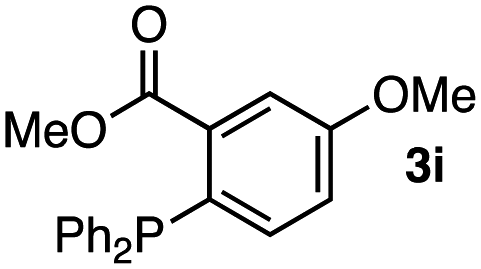
|
Quant. | 0/100 |
| 9 |

|
Quant. | 0/100 |
| 10 |

|
97 | 0/100 |

With an idea for assembling modules through selective azaylide formation, we then examined the stability of azaylides under various conditions (Table 2). Treatment of azaylides 4a, 4b, 5a, and 5b with an aqueous NaOH showed their good stability under basic conditions (entries 1–4), in which a small amount of phosphine oxide 6b was observed when using azaylide 5b prepared from tetrafluorophenyl azide 2a and diphenyl(2-(methoxycarbonylphenyl))phosphine (3b) (entry 4). In the case of hydrolysis of azaylides under acidic conditions with hydrochloric acid, azaylides 4a and 4b prepared from dichlorophenyl azide 1a with triphenylphosphine (3a) and phosphine 3b bearing ortho ester moiety were more stable than azaylides 5a and 5b from tetrafluorophenyl azide 2a (entries 5 and 6 vs. entries 7 and 8). In particular, we found that quantitative cleavage of azaylide 5a took place with aqueous hydrochloric acid (entry 7), and the stability was significantly improved by the presence of ester moiety (entry 8). In addition, no reaction proceeded when azaylides 4a, 4b, 5a, and 5b were treated with (MeCN)4CuBF4 and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA),9 which were frequently used for the copper-catalyzed azide–alkyne cycloaddition (CuAAC) (entries 9–12). These results suggested that azaylides will be stable under the CuAAC conditions.
|
|
||||
|---|---|---|---|---|
| Entry | Azaylide | Conditions | Recovery of 4a, 4b, 5a, or 5b/% | 6, Yield/% |
| 1 | 4a | 1 M NaOH aq., THF, rt | Quant. | 6a, 0 |
| 2 | 4b | 1 M NaOH aq., THF, rt | Quant. | 6b, 0 |
| 3 | 5a | 1 M NaOH aq., THF, rt | Quant. | 6a, 0 |
| 4 | 5b | 1 M NaOH aq., THF, rt | 94 | 6b, 6 |
| 5 | 4a | 1 M HCl aq., THF, rt | 88 | 6a, 12 |
| 6 | 4b | 1 M HCl aq., THF, rt | Quant. | 6b, 0 |
| 7 | 5a | 1 M HCl aq., THF, rt | 0 | 6a, Quant. |
| 8 | 5b | 1 M HCl aq., THF, rt | 77 | 6b, 23 |
| 9 | 4a | (MeCN)4CuBF4 (5 mol%) | Quant. | 6a, 0 |
| TBTA (5 mol%) | ||||
| CH2Cl2, rt | ||||
| 10 | 4b | (MeCN)4CuBF4 (5 mol%) | Quant. | 6b, 0 |
| TBTA (5 mol%) | ||||
| CH2Cl2, rt | ||||
| 11 | 5a | (MeCN)4CuBF4 (5 mol%) | Quant. | 6a, 0 |
| TBTA (5 mol%) | ||||
| CH2Cl2, rt | ||||
| 12 | 5b | (MeCN)4CuBF4 (5 mol%) | Quant. | 6b, 0 |
| TBTA (5 mol%) | ||||
| CH2Cl2, rt | ||||

Selective azaylide formation enabled us to accomplish sequential Staudinger reactions of diazide 9 with phosphines 3a and 3b (Fig. 2). Diazide 9 having dichlorophenyl and tetrafluorophenyl azide moiety was successfully synthesized from mono-Boc-protected diamine 7 (Fig. 3A). Indeed, after preparation of the corresponding acid chloride from carboxylic acid 1b, amide formation took place smoothly to afford 2,6-dichlorophenyl azide 8. Then, deprotection of Boc group with trifluoroacetic acid and acylation with acid chloride 2b resulted in the efficient synthesis of diazide 9 without damaging two azido groups. Treatment of diazide 9 with o-ester-substituted triarylphosphine 3b furnished azaylide 10 quantitatively leaving 2,6-dichlorophenyl azide moiety untouched (Fig. 3B). The second rapid azaylide formation proceeded smoothly to provide bis(iminophosphorane) 11 in high yield. Due to the fast azaylide formation of 2,6-dichlorophenyl azide with triphenylphosphine (kobs = 0.63 M−1 s−1) and good biocompatibility,6 this sequential conjugation would serve in the synthesis of functional molecules and chemical modification of biomolecules.
 | ||
| Fig. 3 (A) Synthesis of diazide 9. (B) Sequential Staudinger reactions. | ||
To showcase the selective azaylide formation, we succeeded in the efficient assembly of modules using newly developed trivalent platform 16 (Fig. 4). Preparation of azide 15 was achieved from azide 12 by N-propargylation, Staudinger reduction, and N-acylation with acid chloride 2b in good yields (Fig. 4A). Then, deprotection of Boc group followed by acylation with acid chloride 1c took place efficiently to afford trivalent platform 16 without damaging two azide moieties and propargyl group. Of note, 16 was easily constructed in modular synthetic manner through the Staudinger reduction of alkyne 14. We achieved selective azaylide formation by treatment of trivalent platform 16 with ester-substituted phosphine 3b followed by Staudinger reaction with triphenylphosphine and subsequent CuAAC reaction resulting in the efficient synthesis of bis(iminophosphorane) 17 in good yields leaving terminal alkyne moiety intact (Fig. 4B). The simple protocol realized a one-pot triple-click assembly, which would enable us to synthesize multifunctional molecules such as probe compounds from easily available modules (Fig. 4C). Moreover, alkyne selective reaction was accomplished by treatment of trivalent platform 16 with picolyl azide 1810 furnishing triazole 19 with tetrafluorophenyl and dichlorophenyl azide moieties (Fig. 4D). Thus, the order of module assembly could be switched by changing reaction partners. Since rapid azaylide formations served in the efficient chemical labeling using living cells,6,7 functionalized azides preparable from 16 would be of great significance in various research fields such as chemical biology.
 | ||
| Fig. 4 (A) Synthesis of 16. (B) Triple-click assembly. (C) One-pot triple click assembly. (D) Alkyne selective reaction. | ||
Assembly of HaloTag ligand, fluorescent coumarin 102, and biotin moieties was realized by efficient click reactions with trivalent platform 16 (Fig. 5). Indeed, selective azaylide formation took place smoothly without decomposing primary alkyl chloride. Then, we accomplished the second azaylide formation with fluorescent phosphine 21 and following CuAAC reaction with biotinyl azide 22 providing adduct 23 leaving reactive functional groups untouched. Therefore, highly functionalized molecular probes will be synthesized in short steps in a modular synthetic method onto trivalent platform 16.
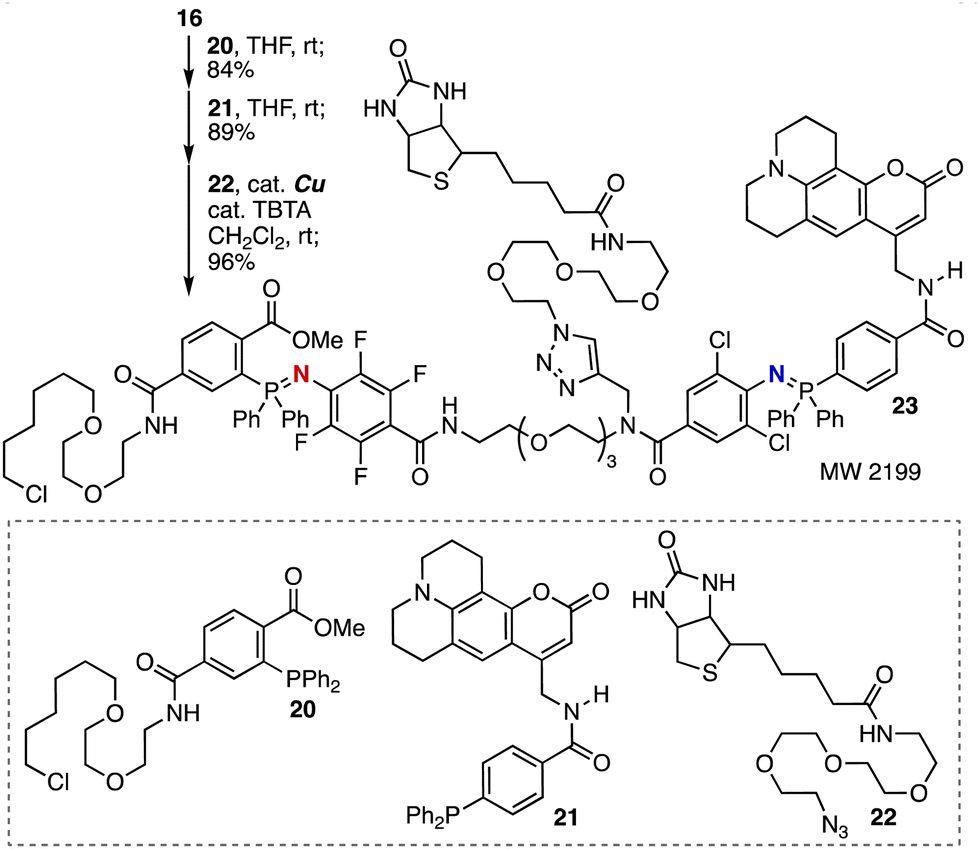 | ||
| Fig. 5 Synthesis of 23 by triple-click assembly. | ||
In summary, we have succeeded in the selective azaylide formation of tetrafluorophenyl azide moiety leaving dichlorophenyl azide side intact. Since rapid azaylide formation of dichlorophenyl azide can be performed using the resulting dichlorophenyl azide and these reactions proceed without damaging an alkyne moiety, the newly designed trivalent platform 16 enabled us to achieve efficient triple click assembly of functional modules. Moreover, since we preliminary found that selective azaylide formations of the tetrafluorophenyl azide moiety can be achieved even in the presence of alkyl azide and dichlorophenyl azide moieties,11 novel trivalent and tetravalent platforms will be developed in future. Further studies including applications to develop novel molecular probes and sequential bioconjugation methods using trivalent platforms are ongoing.
This work was supported by JSPS KAKENHI Grant Number 23K179200 (S.Y.) and Asahi Glass Foundation (S.Y.).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Lahann, Click Chemistry for Biotechnology and Materials Science, John Wiley & Sons, West Sussex, 2009 Search PubMed.
- For reviews of sequential click reactions, see: (a) D. M. Beal and L. H. Jones, Angew. Chem., Int. Ed., 2012, 51, 6320 CrossRef CAS PubMed; (b) A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131 RSC; (c) Z.-J. Zheng, D. Wang, Z. Xu and L.-W. Xu, Beilstein J. Org. Chem., 2015, 11, 2557 CrossRef CAS PubMed; (d) A. Maruani, D. A. Richards and V. Chudasam, Org. Biomol. Chem., 2016, 14, 6165 RSC; (e) S. Yoshida, Org. Biomol. Chem., 2020, 18, 1550 RSC; (f) D. Sato, Z. Wu, H. Fujita and J. S. Lindsey, Organics, 2021, 2, 161 CrossRef CAS.
- For recent our studies on sequential click reactions using tri- or tetravalent platforms, see: (a) T. Meguro, Y. Sakata, T. Morita, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 4720 RSC; (b) N. Terashima, Y. Sakata, T. Meguro, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 14003 RSC; (c) H. Takemura, S. Goto, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 15541 RSC; (d) S. Yoshida, Y. Sakata, Y. Misawa, T. Morita, T. Kuribara, H. Ito, Y. Koike, I. Kii and T. Hosoya, Chem. Commun., 2021, 57, 899 RSC; (e) H. Takemura, G. Orimoto, A. Kobayashi, T. Hosoya and S. Yoshida, Org. Biomol. Chem., 2022, 20, 6007 RSC.
- For recent examples of sequential click reactions using trivalent platforms, see: (a) V. Vaněk, J. Pícha, B. Fabre, M. Buděšínský, M. Lepšík and J. Jiráček, Eur. J. Org. Chem., 2015, 3689 CrossRef; (b) B. Fabre, J. Pícha, V. Vaněk, M. Buděšínský and J. Jiráček, Molecules, 2015, 20, 19310 CrossRef CAS PubMed; (c) B. Fabre, J. Pícha, V. Vaněk, I. Selicharová, M. Chrudinová, M. Collinsová, L. Žáková, M. Buděšínský and J. Jiráček, ACS Comb. Sci., 2016, 18, 710 CrossRef CAS PubMed; (d) A.-C. Knall, M. Hollauf, R. Saf and C. Slugovc, Org. Biomol. Chem., 2016, 14, 10576 RSC; (e) T. Yokoi, H. Tanimoto, T. Ueda, T. Morimoto and K. Kakiuchi, J. Org. Chem., 2018, 83, 12103 CrossRef CAS PubMed; (f) T. Yokoi, T. Ueda, H. Tanimoto, T. Morimoto and K. Kakiuchi, Chem. Commun., 2019, 55, 1891 RSC; (g) K. Maegawa, H. Tanimoto, S. Onishi, T. Tomohiro, T. Morimoto and K. Kakiuchi, Org. Chem. Front., 2021, 8, 5793 RSC.
- (a) T. K. Heiss, R. S. Dorn and J. A. Prescher, Chem. Rev., 2021, 121, 6802 CrossRef CAS PubMed; (b) E. Poulou and C. P. R. Hackenberger, Isr. J. Chem., 2023, 63, e202200057 CrossRef CAS.
- T. Meguro, N. Terashima, H. Ito, Y. Koike, I. Kii, S. Yoshida and T. Hosoya, Chem. Commun., 2018, 54, 7904 RSC.
- M. Sundhoro, S. Jeon, J. Park, O. Ramström and M. Yan, Angew. Chem., Int. Ed., 2017, 56, 12117 CrossRef CAS PubMed.
- (a) W. Luo, J. Luo, V. V. Popik and M. S. Workentin, Bioconjugate Chem., 2019, 30, 1140 CrossRef CAS PubMed; (b) L. Cheng, X. Kang, D. Wang, Y. Gao, L. Yi and Z. Xi, Org. Biomol. Chem., 2019, 17, 5675 RSC; (c) X. Kang, X. Cai, L. Yi and Z. Xi, Chem. – Asian J., 2020, 15, 1420 CrossRef CAS PubMed; (d) H.-Y. Lin, C.-F. Chen, C.-H. Chen, J.-L. Yeh, T.-T. Huang, Y.-C. Chu and C.-C. Chu, ACS Appl. Polym. Mater., 2022, 4, 7518 CrossRef CAS; (e) M. Yamashina, H. Suzuki, N. Kishida, M. Yoshizawa and S. Toyota, Angew. Chem., Int. Ed., 2021, 60, 17915 CrossRef CAS PubMed; (f) A. Blázquez-Martín, S. Bonardd, E. Verde-Sesto, A. Arbe and J. A. Pomposo, ACS Polym. Au, 2024, 4, 140 CrossRef.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (c) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853 CrossRef CAS PubMed.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Angew. Chem., Int. Ed., 2012, 51, 5852 CrossRef CAS PubMed.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: https://doi.org/10.1039/d4cc02723j |
| This journal is © The Royal Society of Chemistry 2024 |