
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetraphenylpentalenide organolanthanide complexes†
Nicholas J.
Katzer‡
 ab,
Mandeep
Kaur‡
c,
Asmita
Sen‡
d,
Rupal
Nimaiyar
a,
Jochen
Autschbach
*d,
Polly L.
Arnold
*ab and
Ulrich
Hintermair
*c
ab,
Mandeep
Kaur‡
c,
Asmita
Sen‡
d,
Rupal
Nimaiyar
a,
Jochen
Autschbach
*d,
Polly L.
Arnold
*ab and
Ulrich
Hintermair
*c
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: pla@berkeley.edu
bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cDepartment of Chemistry, University of Bath, BA2 7AY, UK. E-mail: u.hintermair@bath.ac.uk
dDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260, USA. E-mail: jochena@buffalo.edu
First published on 12th July 2024
Abstract
The D2h symmetrical 1,3,4,6-tetraphenylpentalenide is an excellent ligand for the stabilisation of strongly coloured bis(pentalenide) LnIII sandwich complexes. These easily accessible compounds complement previously reported lanthanide organometallics and provide new opportunities to understand the roles of the f-orbitals in electronic structure and bonding.
Organometallic complexes have made major contributions to the understanding of electronic structure and bonding across the periodic table. Sandwich molecules such as ferrocene and bis(benzene) chromium have led the way and allowed for applications in organic spintronics and single molecule memory storage.1 F-block compounds can display fascinating electronic properties at the quantum level including magnetism, Kondo behaviour, and superconductivity.2–6 Fundamental studies on f-block complexes with unusual electronic structures can help explain these phenomena, potentially providing transformative breakthroughs for quantum computing and future technologies at the nano-scale.7–12 Surprisingly, the range of organometallic ligands with the capacity to form bonding interactions with orbitals of many different symmetries is still small and limited mostly to monocyclic aromatic hydrocarbons such as 6π cyclopentadienide (C5H5−, Cp−)13 and 10π cyclooctatetraenide (C8H82−, COT2−)14 derivatives.15 The use of bicyclic 10π pentalenide (C8H62−, Pn2−; Fig. 1) in f-block organometallic chemistry has grown in popularity due to its D2h symmetry allowing for favorable bonding interactions involving δ-symmetry orbitals and the metal's dz2 which are not available in the COT analogues due to their D8h symmetry.16 Besides the parent Pn2− synthesised by in situ deprotonation of unstable dihydropentalene,17 1,4-disilylated derivatives (Pn†)2− with increased stability and solubility have been reported.18 A hexamethylated pentalenide (Pn*)2− can also be accessed by a multi-step organic synthesis,19 and more recently 1,3,4,6-tetraphenyl pentalenide (Ph4Pn2−) has been reported to be accessible through the ring-closing condensation of Cp′s with enones.20,21
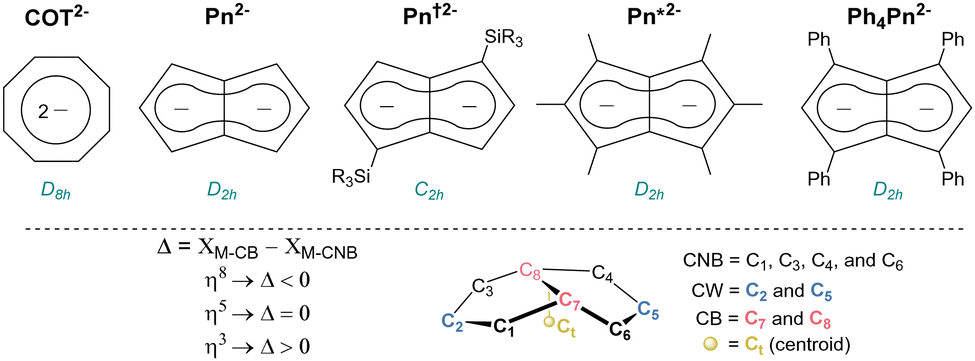 | ||
| Fig. 1 Dianionic 10π ligands for organometallic complexes (upper) and positional numbering and hapticity definition of pentalenides (lower). | ||
Bis(η8-pentalenide) sandwich compounds [M(η8-C8Hn(R)6−n)2] have been reported for Ti, Zr, Hf, Ce, Th, and U.18 These formal 20 electron complexes typically feature a staggered conformation with some degree of folding of the pentalenide around its bridgehead carbons (CB, C7–C8; Fig. 1).22 We were interested in the f-block chemistry of sandwich structures such as K[CeIII(η8-Pn*)]2,23 and MIV (M = Ce, U, Th) containing [M(η8-Pn†)]2 (Fig. 2),24–26 because the ground state electronic structure of cerocene, and also the actinocenes, has been the subject of much debate.27 In particular, comparison of the planar D8h symmetrical COT2− with the folded D2h symmetrical Pn2− in the stabilisation of f-block cations should offer further insight into the covalency in f-block bonding.28–30 The ground state electronic structure of [Ce(COT)2] is probably best viewed as a mixture of ∼80% CeIII with an isolated 4f1 cation coupled to an unpaired electron in the rings' π orbitals, and ∼20% of a state containing the CeIV ion sandwiched by two aromatic 10π dianions with significant Ce 4f–ring covalency.31–33 Pentalenides have also been shown to support pianostool f-block complexes with interesting properties. For example, [(η8-Pn†)DyIII(η5-Cp*)] behaves as a single molecule magnet with an energy barrier of 245 cm−1.34 In related UIII complexes the fold of the Pn2− ligand was noted as important for subsequent reactivity such as dinitrogen reduction to form (N2)[U(η5-Cp*)(η8-Pn†)]2.35,36
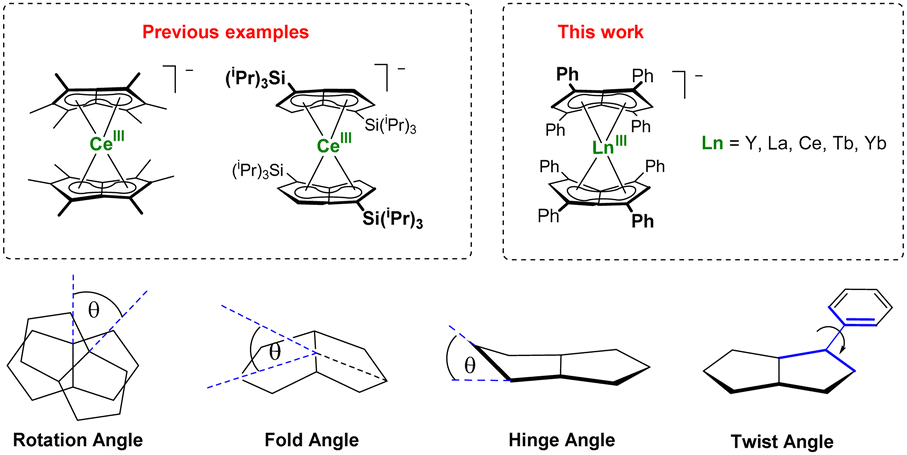 | ||
| Fig. 2 F-block bis(pentalenide) sandwich complexes (upper) and geometric definitions of relevant structural parameters (lower). | ||
Here we explore the organometallic lanthanide chemistry of 1,3,4,6-tetraphenylpentalenide Ph4Pn2− with a range of 4f cations and study the metal–ligand bonding both experimentally and computationally.
Salt metathesis reactions between dilithium, disodium, or dipotassium salts of Ph4Pn2− reacted cleanly with half an equivalent of anhydrous lanthanide(III) halides in THF or DME to afford the pentalenide Ln sandwich ‘ate’ complexes [M(sol)m][LnIII(Ph4Pn)2]2 (1-Ln) for Ln = Y (orange), La (bright orange), Ce (rust-orange), Tb (bright red), Yb (red-orange) (Scheme 1). These are a good representation of the size and Lewis acidity range for the rare earths, with rcovalent(6-coordinate LnIII) = 0.900 Å (Y), 1.032 Å (La), 1.01 Å (Ce), 0.923 Å (Tb), 0.868 (Yb).37 The reactions proceed most cleanly for the largest Ln, with isolated yields of 76% for 1-La, 87% for 1-Ce, and 77% for 1-Tb, but only a few single crystals could be isolated from the reaction that afforded 1-Yb. 1H NMR spectroscopic analysis of the crude product solution of the latter suggested the formation of a mixture of products, so further efforts to isolate pure material were not pursued. We have also crystallographically characterized the new DME adducts of Li2[Ph4Pn], K2[Ph4Pn], and 1-Ln (Fig. S12 and S22, ESI†).
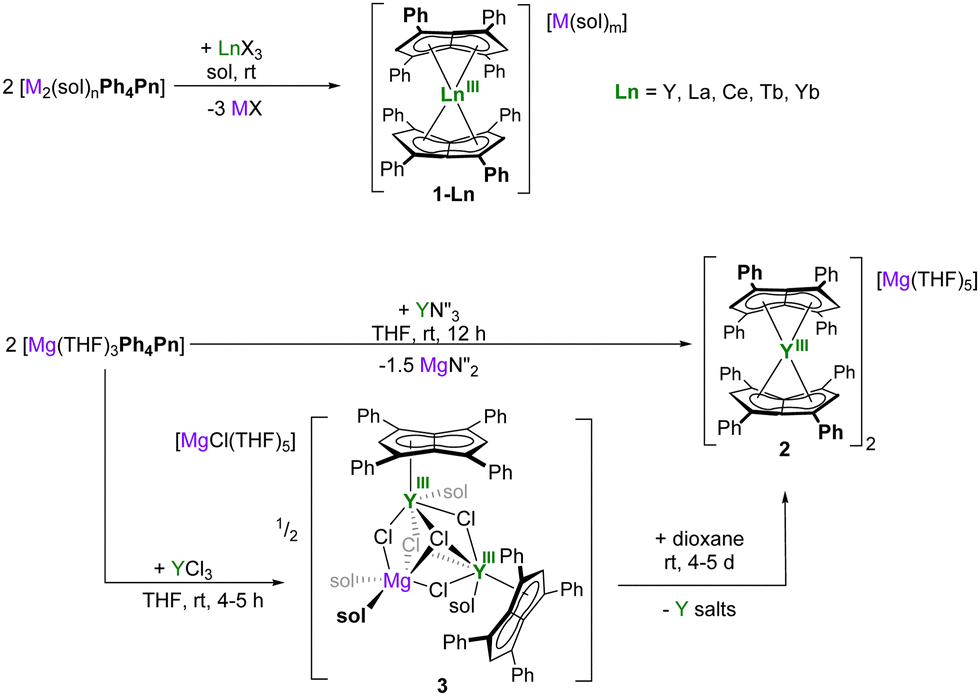 | ||
| Scheme 1 Synthesis of [M(sol)m][LnIII(Ph4Pn)2]2 (1-Ln) for Ln = Y, La, Ce, Tb, Yb from group 1 pentalenide salts (for Ln = Y, M = Li, n = 4, X = Cl, sol = THF, M(sol)m = Li(DME)3; for Ln = Ce, M = K, n = 2, X = Br, sol = DME, M(sol)m = K(DME)4; for Ln = Yb, M = Na, n = 6, X = Cl, sol = THF, M(sol)m = Na(THF)5), synthesis of the Mg analogue of 1-Y, [Mg(THF)5][YIII(Ph4Pn)2]2 (2) from a magnesium pentalenide, and a half-sandwich cluster [MgCl(THF)5][(YIIIPh4Pn)2(μ-Cl)5Mg(THF)4] (3). | ||
Magnesium pentalenides may also be used to make rare earth pentalenide sandwich complexes.38 A metathesis reaction between Mg[Ph4Pn] and Y(N′′)3 (N′′ = bis{trimethylsilyl}amide, Scheme 1) gave the magnesium congener of 1-Y, orange [Mg(THF)5][YIII(Ph4Pn)2]2 (2) in 40% isolated yield plus Mg(N′′)2 as a by-product. An orange slurry of equimolar YCl3 and Mg[Ph4Pn] in THF at room temperature immediately turns red, and work-up after four hours yields the trimetallic [(MgII)2YIII] halide bridged complex [MgCl(THF)5][(YIIIPh4Pn)2(μ-Cl)5Mg(THF)4] (3, Scheme 1). The same reaction with group 1 pentalenide salts gave similar results (Fig. S1, ESI†), suggesting the formation of halide-bridged mono-pentalenide clusters to be characteristic of the harder YIII cation.39 Attempts to break up the halide bridges to access monomeric pianostool YIII complexes by addition of AgPF6, B(C6F5)3, or AlCl3 led to decomposition into unidentifiable mixtures, but 3 could be converted to the bis(pentalenide) sandwich [Mg(THF)5][YIII(Ph4Pn)2]2 (2) congener to 1-Y by addition of dioxane, as confirmed by NMR and XRD (Fig. S2, ESI†). All these reactions support the expectation that the bis-η8 sandwich complexes are the thermodynamically favored products for the rare earth cations.
The 1H NMR spectra of the diamagnetic 1-Ln and 2 show diagnostic resonances for the pentalenide wingtip protons Hw which are sensitive reporters for the degree of shielding of the 10π system (Fig. 1).40 Spectra of 1-La in THF show a single resonance for the two equivalent Hw at 6.39 ppm and a corresponding Cw resonance at 122.5 ppm. Comparatively, the NMR spectra of 2 in THF contained a single 1H resonance for Hw at 7.20 ppm and a 13C resonance for Cw at 124.1 ppm. The latter is 8.6 ppm higher frequency than in Mg[Ph4Pn] which exists as a solvent-separated ion pair in ethereal solution.38 The NMR spectra of the cluster complex 3 also show a D2h symmetrical pentalenide with one sharp set of equivalent Hw at temperatures down to –60 °C in THF, while DOSY analysis suggests the cluster anion of 3 remains intact in solution (D = 6.325 × 10−10 m2 s−1 corresponding to 1462 g mol−1 compared to MW = 1484 g mol−1; Fig. S3, ESI†). These observations support the presence of strong Mg–Cl–Y interactions in the trinuclear cluster with rapid ring slippage of the η5 pentalenide on YIII.41
The D2h symmetry of 1,3,4,6-Ph4Pn2− greatly facilitates structural analysis of the new sandwich complexes in solution and in the solid state, as it eliminates the formation of meso, rac, and twist isomers seen with the C2h symmetrical 1,4-bis(silyl)-substituted (Pn†)2− (Fig. 1).181-Ln crystallize as either solvent-separated ion pairs (SSIP) or contact ion pairs (CIP) depending on the solvent and countercation. In each case, the LnIII ion sits on a crystallographic C2 axis positioned centrally between two parallel (Ct–Ln–Ct′ angles close to 180°) but staggered Ph4Pn2− ligands which are rotated by 38–76° against each other with noticeable inward folding by ∼24° to form a hydrocarbon capsule around the metal.24 Each C5 ring of the pentalenide remains planar with no noticeable wingtip hinging, and phenyl twist angles are in the typical range of 19–49° previously observed for other s- and d-block complexes of Ph4Pn2−.20,38,41 Ring slippage values of Δ = −0.3 indicate bis-η8 coordination for all metals and M–Ct distances are similar once normalized for the metal radius, except for 1-Yb where the M–Ct of 2.280 Å is anomalously long, and 2 where the M–Ct of 2.048 Å is anomalously short. Both of these also have larger pentalenide rotation angles of 76.2 and 71.9° respectively, compared to an average of 38.6° for the others. 1-Ce(DME) is shown in Fig. 3 (left) as an example. The solid-state structure of complex 3 (Fig. 3, right) can be considered to comprise two [YIII(Ph4Pn)Cl2]− anions linked by a [MgCl]+ cation through bridging halides, with THF molecules saturating the coordination spheres of MgII and YIII. In contrast to 1 and 2, in 3 the Ph4Pn2− ligands bind to each YIII in nearly perfect η5 coordination (Δ = −0.006). The 2.319 Å Y–Ct distance in 3 is significantly longer than that in 2 (2.048 Å), consistent with lower hapticity and thus weaker binding.
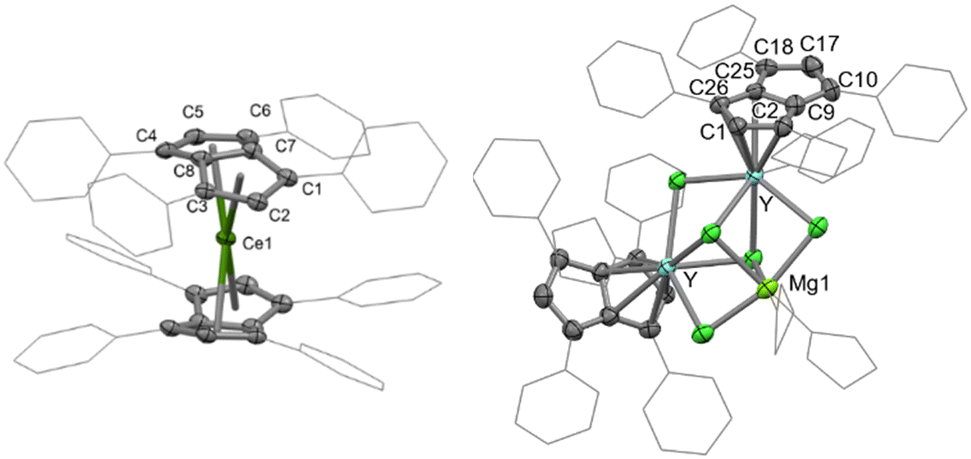 | ||
| Fig. 3 Molecular structures of (left) the anion of 1-Ce(DME) and (right) the anionic MgY2 pentalenide cluster of 3. Selected ellipsoids drawn at 50% probability; H atoms, counter cations and lattice solvent omitted for clarity. | ||
Starting from the crystal structure data, molecular geometries were optimised computationally for [LnIII(η8-Ph4Pn)2]− with Ln = La, Ce, and Tb in their respective electronic ground states with S = 0, ½, 3 using the PBE0 functional (see ESI† for details).42–44 All structures were confirmed as minima via harmonic vibrational frequency calculations, and all Ln–C distances agreed very well with those found experimentally (Fig. S23–S25, S30 and Tables S1–S3, S9–S11, ESI†). Superimposing the optimized geometries on the crystal structures showed the latter to be representative of what would likely be found in solution (as obtained from the calculations; see Fig. S26 and Table S4, ESI†). The chemical bonding and metal electronic configurations were analysed by Natural Localized Molecular Orbitals (NLMOs) and Natural Population Analysis (NPA). Each ligand has a 10-electron 5-orbital π system in the bicyclic core represented by ligand-centred NLMOs that are strongly delocalized over the pentalenide (as is typical for delocalized π-donor ligands)40 and clearly donating electron density to the metal. For example, the ten (both ligands) relevant 1-La NLMOs each contain 3–5% La character, which sums to about 0.24/0.57 electrons being donated to 4f/5d and the remainder into more diffuse shells (Table S5, ESI†). At La, these NLMOs are mainly of 5d character (>50%) with secondary 4f admixtures (Fig. S27, ESI†). The La charge as determined by NPA is +1.85, smaller than the formal +3 charge, as expected because of ligand to metal electron donation. Optimised molecular structural data in comparison with experimental parameters are provided in Tables S1–S3 (ESI†), along with Ln–C Wiberg bond orders (WBOs). In agreement with a recent suggestion,45 the bridgehead carbons, which are closest to the metal, generate the largest WBOs. Similar bonding patterns were found in 1-Ce and 1-Tb (Table S5 and Fig. S24–S25, S28 and S29, ESI†) but we note that there is almost no donation into 4f for the Tb system, with the occupancy determined as 4f0.24, 4f1.22, and 4f8.04 for La, Ce, and Tb congeners respectively. The calculations also reveal π-delocalization between the pentalenide core and the phenyl substituents as found earlier for the THF-solvated Li salts,40 resulting in reduced ligand-to-metal donation compared to the parent Pn2− (Table S6, ESI†). The overall −1 charge of each complex is balanced by the donation between ligands and the metal, with the eight phenyl groups in each complex collectively holding a charge of about −0.5 (Table S7, ESI†). We also performed comparative calculations for the analogous [LnIII(η8-COT)2]− complexes (Table S8, ESI†) which show overall the order of ligand-to-metal donation increases from Ph4Pn2− through COT2− to Pn2−, with most of the differences impacting the donation into 5d and more diffuse shells. The extent of donation into 4f is similar for the three types of ligands.
The experimental UV-vis-NIR spectra of 1-La, 1-Ce and 1-Tb in DME are dominated by an intense absorption around 380 nm with extinction coefficients of >30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1cm−1 (Fig. 4 and Fig. S10, ESI†), whereas for the YIII complexes 2 and 3 in THF these transitions are slightly less intense and blue-shifted by around 30 nm (Fig. S11, ESI†). The spectra of 1-La and 1-Ce further feature weaker lower-energy transitions around 550 and 750 nm.
000 M−1cm−1 (Fig. 4 and Fig. S10, ESI†), whereas for the YIII complexes 2 and 3 in THF these transitions are slightly less intense and blue-shifted by around 30 nm (Fig. S11, ESI†). The spectra of 1-La and 1-Ce further feature weaker lower-energy transitions around 550 and 750 nm.
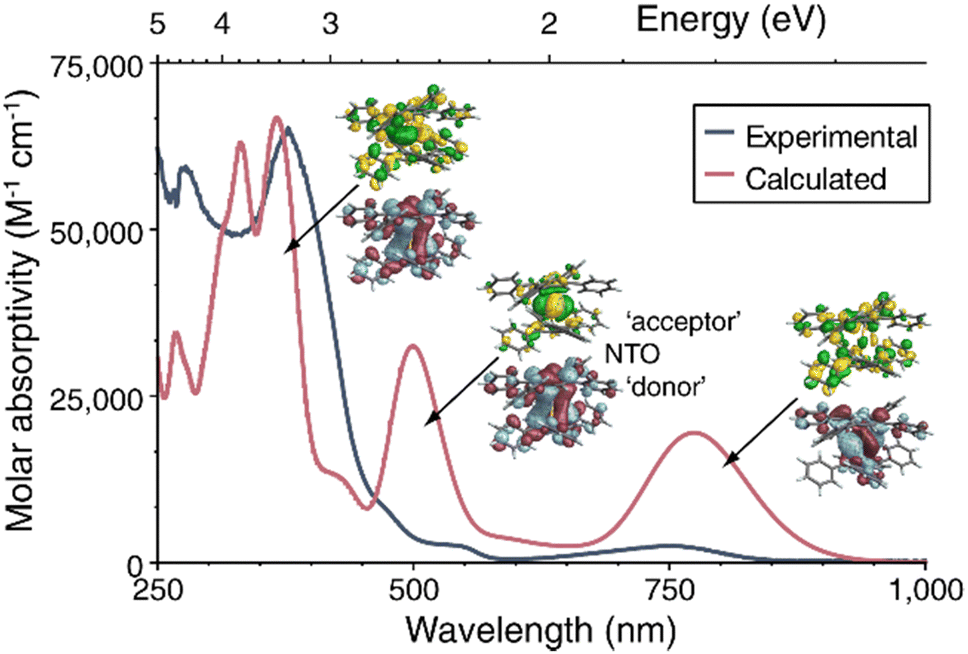 | ||
| Fig. 4 UV-vis-NIR spectrum of 1-La in DME at room temperature (dark blue). TD-DFT calculated spectrum (red). Insets: representative NTOs (isosurfaces) for the most intense transitions. | ||
Time-dependent DFT calculations of the electronic excitation spectra of 1-Ln reproduced the observed spectral patterns in gas phase calculations as well as with the use of a solvent model, although at the chosen level of broadening the computed bands are more intense in the low-energy regions than the experimental spectra (Fig. S31, S33 and S36, ESI†). The natural transition orbital (NTO) analysis of 1-La (Fig. 4 and Fig. S32, ESI†) shows low-energy bands around 1.6 eV/780 nm and 2.5 eV/500 nm. The calculated spectrum also features comparatively intense bands at energies of 3.1 eV and higher/400 nm and below. According to the NTO analysis, an intense transition calculated around 375 nm has mixed intra-ligand, MLCT, and metal 5d to 6p character, the latter being possible because of the donation into 5d in the ground state. The most intense transition in the 500 nm peak is mixed LMCT and 5d–5d, with the acceptor NTO of the dominant contribution to the transition having approximate δ symmetry about the Ct–Ln–Ct′ axis. The donor (‘hole’) NTO in both cases corresponds closely to the HOMO−1. The band at lowest energy is assigned as HOMO to ligand π* with weak metal contributions in the acceptor NTO (Fig. 4). The peak assignment is qualitatively similar for the corresponding absorptions of 1-Ce (Fig. S33–S35, ESI†) and 1-Tb (Fig. S36–S37, ESI†). In contrast to the analogous COT2− complexes, participation of the phenyl substituents is clearly noticeable in the NTOs. Furthermore, the pentalenides induce mixing of rotational symmetries of σ, π, δ, and φ where the metal and ligand fragment orbitals are similar in energy (Fig. 4 and Fig. S38–S43, ESI†).
In conclusion, the Ph4Pn2− ligand is highly suited for stabilising f-block organometallics giving access to both half-sandwich as well as sandwich complexes of the rare earths. Its ease of synthesis, high symmetry and ready crystallisation facilitate NMR and XRD analysis and promises access to the first homologous series of f-block pentalenide sandwich complexes to investigate fundamental aspects of electronic structure and bonding. In comparison to unsubstituted Pn2− and COT2− we find Ph4Pn2− to be a better acceptor, i.e. the order of ligand-to-metal electron donation (primarily into 5d orbitals) is Pn > COT > Ph4Pn. This will prove useful for the stabilisation of electron-rich f-block organometallics where the comparatively low symmetry offers positive overlap of ligand frontier orbitals with the metal ion's dz2 orbital which is not possible in the COT2− complexes.
The synthetic parts of this research were supported by UK's Royal Society (awards URF\R\221049 and RF\ERE\221083), the Heavy Element Chemistry Program and the Molecular Qubits Project of the Quantum Information Science program of the U.S. DOE, Office of Science, Basic Energy Sciences, CSGB Division under contract DE-AC02-05CH11231 (PLA). MK and UH thank Dr Gabriele Kociok-Köhn from the Chemical Characterisation Analysis at Bath for XRD analyses. JA acknowledges DOE grant DE-SC0001136 (Heavy Element Chemistry) for supporting the theoretical component of this study. We thank Drs. Hasan Celik, Raynald Giovine, and Pines Magnetic Resonance Center's Core NMR Facility for spectroscopic assistance. One instrument used in this work was in part supported by NIH S10OD024998 and the PMRC Core. Computations were carried out at the Center for Computational Research at the University at Buffalo (https://hdl.handle.net/10477/79221).
Data availability
Crystallographic datasets are available from the CCDC deposition numbers 2332849, 2357823–2357832 and other analytical data can be retrieved via DOI: https://doi.org/10.5061/dryad.sxksn03bf.Conflicts of interest
There are no conflicts of interest to declare.References
- H. Werner, Angew. Chem., Int. Ed., 2012, 51, 6052 CrossRef CAS PubMed.
- S. Bart and E. Schelter, Organometallics, 2017, 36, 4507 CrossRef CAS.
- L. Ungur and L. Chibotaru, Inorg. Chem., 2016, 55, 10043 CrossRef CAS PubMed.
- C. Gould, et al. , J. Am. Chem. Soc., 2019, 141, 12967 CrossRef CAS PubMed.
- N. Mahieu, et al. , Chem. Sci., 2023, 14, 443 RSC.
- E. Castellanos and S. Demir, Inorg. Chem., 2023, 62, 2095 CrossRef CAS PubMed.
- L. Münzfeld, et al. , Angew. Chem., Int. Ed., 2023, 62, e202218107 CrossRef PubMed.
- P. Arnold, et al. , Nat. Chem., 2012, 4, 668 CrossRef CAS PubMed.
- W. Huang, et al. , Nat. Commun., 2013, 4, 1448 CrossRef PubMed.
- S. Minasian, et al. , Chem. Sci., 2014, 5, 351 RSC.
- D. Smiles, et al. , Chem. Sci., 2020, 11, 2796 RSC.
- C. Gould, et al. , Nat. Chem., 2021, 13, 1001 CrossRef CAS PubMed.
- P. Jutzi and N. Burford, Chem. Rev., 1999, 99, 969 CrossRef CAS PubMed.
- F. Sroor, J. Organomet. Chem., 2021, 948, 121878 CrossRef CAS.
- P. Arnold, et al. , Inorg. Chem., 2021, 60, 4162 CrossRef CAS PubMed.
- R. King, Appl. Organomet. Chem., 2003, 17, 393 CrossRef CAS.
- T. Katz and N. Acton, J. Am. Chem. Soc., 1972, 94, 3281 CrossRef CAS.
- F. Cloke, et al. , Coord. Chem. Rev., 2017, 344, 238 CrossRef CAS.
- A. Ashley, A. Cowley and D. O'Hare, Chem. Commun., 2007, 1512 RSC.
- S. Boyt, et al. , Organometallics, 2022, 41, 211 CrossRef CAS.
- N. Jenek, et al. , J. Org. Chem., 2022, 87, 13790 CrossRef CAS PubMed.
- K. Costuas and J.-Y. Saillard, Chem. Commun., 1998, 2047 RSC.
- A. Ashley, et al. , Chem. Commun., 2007, 1515 RSC.
- G. Balazs, et al. , Organometallics, 2007, 26, 3111 CrossRef CAS.
- O. Summerscales and F. Cloke, Coord. Chem. Rev., 2006, 250, 1122 CrossRef CAS.
- F. Cloke and P. Hitchcock, J. Am. Chem. Soc., 1997, 119, 7899 CrossRef.
- O. Walter in Comprehensive Organometallic Chemistry IV, Elsevier, 2022, p. 582 Search PubMed.
- M. Neidig, D. Clark and R. Martin, Coord. Chem. Rev., 2013, 257, 394 CrossRef CAS.
- A. Kerridge, Chem. Commun., 2017, 53, 6685 RSC.
- S. Cooper and N. Kaltsoyannis, Dalton Trans., 2021, 50, 1478 RSC.
- D.-C. Sergentu, et al. , Chem. Eur. J., 2021, 27, 7239 CrossRef CAS PubMed.
- O. Mooßen and M. Dolg, Chem. Phys. Lett., 2014, 594, 47 CrossRef.
- G. Ganguly, et al. , Chem. Eur. J., 2020, 26, 1776 CrossRef CAS PubMed.
- A. Kilpatrick, et al. , Chem. Commun., 2018, 54(51), 7085 RSC.
- F. Cloke and P. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352 CrossRef CAS PubMed.
- N. Tsoureas, et al. , J. Am. Chem. Soc., 2020, 142, 89 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751 CrossRef.
- H. Sanderson, et al. , Inorg. Chem., 2023, 62, 15983 CrossRef CAS PubMed.
- R. Cooper, et al. , Organometallics, 2013, 32, 2228 CrossRef CAS.
- N. Jenek, et al. , Chem. Sci., 2024 10.1039/D3SC04622B.
- H. Sanderson, et al. , Dalton Trans., 2024, 53, 5881 RSC.
- E. Glendening, et al. , Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 142 Search PubMed.
- C. Adamo and V. Barone, Chem. Phys. Lett., 1998, 298, 113 CrossRef CAS.
- E. Baerends, et al., Amsterdam Density Functional (ADF) program, Version 2023.104 Search PubMed.
- E. Reinhart, et al. , Organometallics, 2024, 43, 284 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 2332849, 2357823–2357832. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02570a |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2024 |