
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluoride recovery in degradable fluorinated polyesters†
Christoph
Fornacon-Wood
 a,
Merlin R.
Stühler
b,
Alexandre
Millanvois
b,
Luca
Steiner
b,
Christiane
Weimann
c,
Dorothee
Silbernagl
c,
Heinz
Sturm
c,
Beate
Paulus
b and
Alex J.
Plajer
*a
a,
Merlin R.
Stühler
b,
Alexandre
Millanvois
b,
Luca
Steiner
b,
Christiane
Weimann
c,
Dorothee
Silbernagl
c,
Heinz
Sturm
c,
Beate
Paulus
b and
Alex J.
Plajer
*a
aMakromolekulare Chemie 1, Universität Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: alex.plajer@uni-bayreuth.de
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Arminallee 22, 14195 Berlin, Germany
cBundesanstalt für Materialforschung und -Prüfung (BAM), Unter den Eichen 87, Berlin 12205, Germany
First published on 25th June 2024
Abstract
We report a new class of degradable fluorinated polymers through the copolymerization of tetrafluorophthalic anhydride and propylene oxide or trifluoropropylene oxide which show up to 20 times quicker degradation than the non-fluorinated equivalents and allow for fluoride recovery.
Fluorinated polymers are not only popular materials in a wide range of consumer applications but are currently irreplaceable in many industries.1,2 The low polarizability of the fluorine groups for example renders these polymers more hydrophobic and less adhesive than their non-fluorinated counterparts making them useful as, for example, water repellent and low-friction surface coatings.3–5 Such polymers can be classified as per- and polyfluoroalkyl substances (PFAS) which have come under much scrutiny for being “forever chemicals”, meaning they do not show appreciable degradation in the environment with respect to their time in use.6,7 The majority of fluorinated and semi-fluorinated polymers currently end up in landfill which can result in leakage of microplastics into the surrounding environment, further exacerbating the issue.8,9 This has caused PFAS to be found everywhere from in household pets to Antarctic ice.10,11 For this reason, the design of future fluoropolymers should include viable recycling methods which allow for chemical recyclability so that the materials can be transformed into chemicals that can be easily disposed of, used in other industries or be incorporated into a circular polymer economy;12–14 these considerations must already be taken into account when designing new polymer structures. The fluorine in fluoropolymers is initially mined as fluorite (CaF2) and converted to HF which is then used for the synthesis of a wide range of fluorinated materials (see Fig. 1(a)).15,16
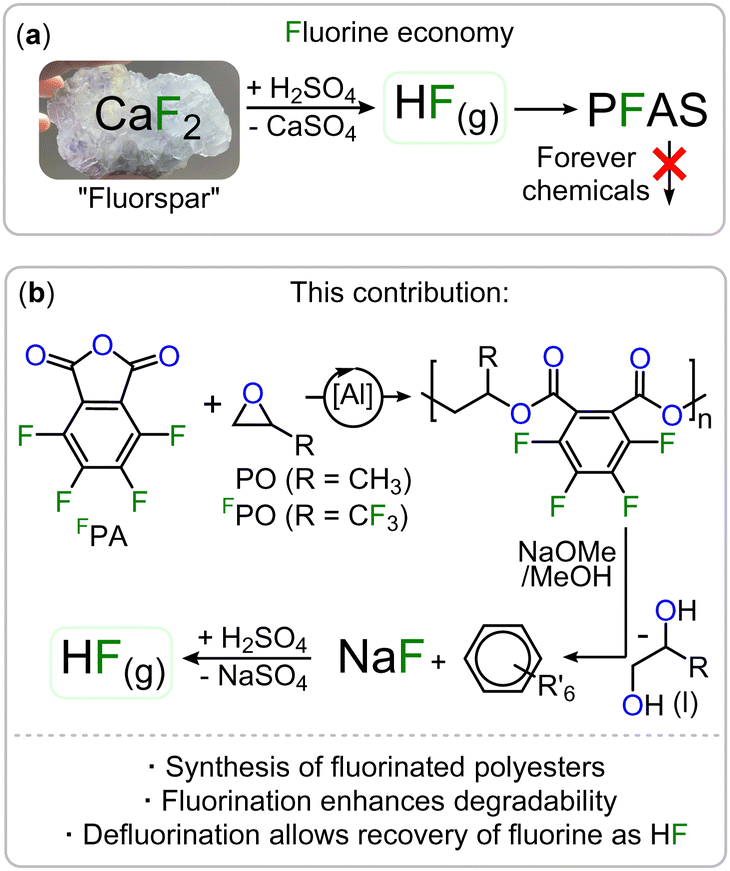 | ||
| Fig. 1 (a) Current fluorine economy resulting in accumulating PFAS. (b) Fluorinated Polyester with in-built recovery options for fluorine. R′ = OMe, COONa, COOMe. | ||
Consequently, another major sustainability issue with our current fluorine economy is that the fluorine bound in these is then not recovered into a useful form.17 In other words, fluorine is a limited resource which will ultimately become scarce and expensive.13 A circular economy should therefore also include methods of extracting fluorine from fluorinated waste into a form where it can be reused. In the general context of moving towards a more circular polymer economy, an increasingly popular approach is the incorporation of ester bonds into the polymer main chain which facilitates degradation and enables chemical recycling methods.18–24 However fluorinated polyesters remain rare.25–27 One approach to synthesise polyesters is the ring-opening copolymerisation (ROCOP) of an epoxide and a cyclic anhydride which has yet to be applied to the synthesis of degradable fluorinated polymers.28–35 As the ROCOP of epoxides with cyclic anhydrides is most robust with phthalic anhydride (PA) we hypothesized that fluorinated variants of these monomers (i.e. tetrafluorophthalic anhydride, FPA see Fig. 1(b)) would give easy access to fluorinated polyesters which we report in this contribution. In order to facilitate the ROCOP of FPA with epoxides, we turned to bicomponent catalysts comprising of a LAl(III)Cl/PPNCl catalyst pair (PPN = Ph3PNPPh3) in which L is a bisphenoxy imine ligand. Here aluminium(III) bound alkoxides from epoxide ring opening insert into cyclic anhydrides to generate carboxylates that dissociate from the Al(III) to vacate a coordination site for epoxides (see Fig. 2(a)). These are then attacked by the carboxylate chain end to close the catalytic cycle, having elongated the polymer chain by two ester bonds.36,37 Attempting FPA/propylene oxide (PO) ROCOP with the LAl(III)Cl (C1–C4, Fig. 2(a), Notes S1, ESI†)/PPNCl catalyst pairs at an initial loading of 1 LAl(III)Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PPNCl:500 PO:550 FPA at 80 °C for 90 min indeed resulted in highly viscous mixtures. 19F NMR spectroscopy reveals de-symmetrisation of the initially symmetric FPA signals into broadened resonances at −136 and −148 ppm (see Fig. 2(b)). GPC in THF relative to a polystyrene standard shows apparent molecular weights of 11.5–17.0 kg mol−1 (Đ = 1.1–1.3). 1H NMR shows that the polymer comprises of 92% ester links with 8% ether errors from PO homo-polymerisation (see Fig. S3, ESI†). Compared to the non-fluorinated case (namely PA/PO ROCOP) FPA/PO ROCOP shows faster polymerisation rates irrespective of the ligand (C1–C4, Fig. 2(a)) complexing aluminium(III) (TOF = 260–311 h−1 for FPA/PO vs. TOF = 48–209 h−1, see ESI† Table S1). Interestingly, while in the latter non-fluorinated case a strong dependence on the electronic nature of the Al-catalyst and therefore its Lewis acidity is observed, in the fluorinated case the same rate is observed within error irrespective of the catalysts’ Lewis acidity (see ESI† Notes S7). We infer that this is due to the electron-withdrawing nature of the fluorine substituents, causing carboxylate chain ends from FPA ring opening to be less coordinating so that de-coordination of these off the Lewis acidic Al(III) centre occurs more readily. This is required prior to the rate determining PO insertion and this also helps explain the rate enhancement observed for FPA. FPA can also be copolymerised with the fluorinated epoxide trifluoropropylene oxide (FPO) to obtain even more fluorinated polyesters. FPO/FPA ROCOP at 1 C4:1 PPNCl:500 FPO:550 FPA yields perfect polyester without ether errors at a Mn = 16.5 (Đ = 1.3) (see ESI† Section S19). Terpolymerising mixtures of FPA and PA either with PO or FPO results in block polymer formation via exclusive FPA ROCOP until full FPA consumption which is followed by PA/PO ROCOP as established by aliquot monitoring (see Fig. S32, ESI†). Similarly, when combining FPA, PA, PO and FPO in one pot, the fluorinated anhydride forms a statistical terpolymer in which PO is incorporated preferentially over FPO in a 7:3 ratio (see Fig. S59, ESI†). This implies that FPA must insert into alkoxide intermediates orders of magnitude faster than PA and this is a likely consequence of the increased electrophilicity due to the electron withdrawing nature of the fluorine substituents, suggesting that these might affect further reactivity of the material (vide infra). To study the effect of fluorination on material properties, we turned to atomic force microscopy force–distance curves (AFM FDC) of polymer films on surface coated silicon wafers (see ESI† Notes S10).38,39 This was necessary due to the brittle nature of the materials preventing investigations of the bulk mechanical properties as samples shattered easily during handling. Polymers with similar apparent molecular weights namely FPA-co-FPO at Mn = 16.5 kg mol−1, FPA-co-PO at Mn = 17.0 kg mol−1 and PA-co-PO at Mn = 17.8 kg mol−1 were employed. Due to weaker chain-chain interaction upon fluorination the material is more prone to irreversible plastic deformation (Dpla(FPA-co-FPO) > Dpla(FPA-co-PO) > Dpla(PA-co-PO)). This agrees with the measured elastic modulus (E(FPA-co-FPO) < E (FPA-co-PO) < E (PA-co-PO)). Recording the approach of the AFM cantilever, a decrease of measured attractive force between AFM Tip and sample surface from 50 nN (PA-co-PO), to 41 nN (FPA-co-PO) to 38 nN (FPA-co-FPO) was found which we infer to be a consequence of reduced surface van der Waals forces.40 Hence fluorination renders the material less adhesive, a typical property benefit for fluorinated polymers.3
:1 PPNCl:500 PO:550 FPA at 80 °C for 90 min indeed resulted in highly viscous mixtures. 19F NMR spectroscopy reveals de-symmetrisation of the initially symmetric FPA signals into broadened resonances at −136 and −148 ppm (see Fig. 2(b)). GPC in THF relative to a polystyrene standard shows apparent molecular weights of 11.5–17.0 kg mol−1 (Đ = 1.1–1.3). 1H NMR shows that the polymer comprises of 92% ester links with 8% ether errors from PO homo-polymerisation (see Fig. S3, ESI†). Compared to the non-fluorinated case (namely PA/PO ROCOP) FPA/PO ROCOP shows faster polymerisation rates irrespective of the ligand (C1–C4, Fig. 2(a)) complexing aluminium(III) (TOF = 260–311 h−1 for FPA/PO vs. TOF = 48–209 h−1, see ESI† Table S1). Interestingly, while in the latter non-fluorinated case a strong dependence on the electronic nature of the Al-catalyst and therefore its Lewis acidity is observed, in the fluorinated case the same rate is observed within error irrespective of the catalysts’ Lewis acidity (see ESI† Notes S7). We infer that this is due to the electron-withdrawing nature of the fluorine substituents, causing carboxylate chain ends from FPA ring opening to be less coordinating so that de-coordination of these off the Lewis acidic Al(III) centre occurs more readily. This is required prior to the rate determining PO insertion and this also helps explain the rate enhancement observed for FPA. FPA can also be copolymerised with the fluorinated epoxide trifluoropropylene oxide (FPO) to obtain even more fluorinated polyesters. FPO/FPA ROCOP at 1 C4:1 PPNCl:500 FPO:550 FPA yields perfect polyester without ether errors at a Mn = 16.5 (Đ = 1.3) (see ESI† Section S19). Terpolymerising mixtures of FPA and PA either with PO or FPO results in block polymer formation via exclusive FPA ROCOP until full FPA consumption which is followed by PA/PO ROCOP as established by aliquot monitoring (see Fig. S32, ESI†). Similarly, when combining FPA, PA, PO and FPO in one pot, the fluorinated anhydride forms a statistical terpolymer in which PO is incorporated preferentially over FPO in a 7:3 ratio (see Fig. S59, ESI†). This implies that FPA must insert into alkoxide intermediates orders of magnitude faster than PA and this is a likely consequence of the increased electrophilicity due to the electron withdrawing nature of the fluorine substituents, suggesting that these might affect further reactivity of the material (vide infra). To study the effect of fluorination on material properties, we turned to atomic force microscopy force–distance curves (AFM FDC) of polymer films on surface coated silicon wafers (see ESI† Notes S10).38,39 This was necessary due to the brittle nature of the materials preventing investigations of the bulk mechanical properties as samples shattered easily during handling. Polymers with similar apparent molecular weights namely FPA-co-FPO at Mn = 16.5 kg mol−1, FPA-co-PO at Mn = 17.0 kg mol−1 and PA-co-PO at Mn = 17.8 kg mol−1 were employed. Due to weaker chain-chain interaction upon fluorination the material is more prone to irreversible plastic deformation (Dpla(FPA-co-FPO) > Dpla(FPA-co-PO) > Dpla(PA-co-PO)). This agrees with the measured elastic modulus (E(FPA-co-FPO) < E (FPA-co-PO) < E (PA-co-PO)). Recording the approach of the AFM cantilever, a decrease of measured attractive force between AFM Tip and sample surface from 50 nN (PA-co-PO), to 41 nN (FPA-co-PO) to 38 nN (FPA-co-FPO) was found which we infer to be a consequence of reduced surface van der Waals forces.40 Hence fluorination renders the material less adhesive, a typical property benefit for fluorinated polymers.3
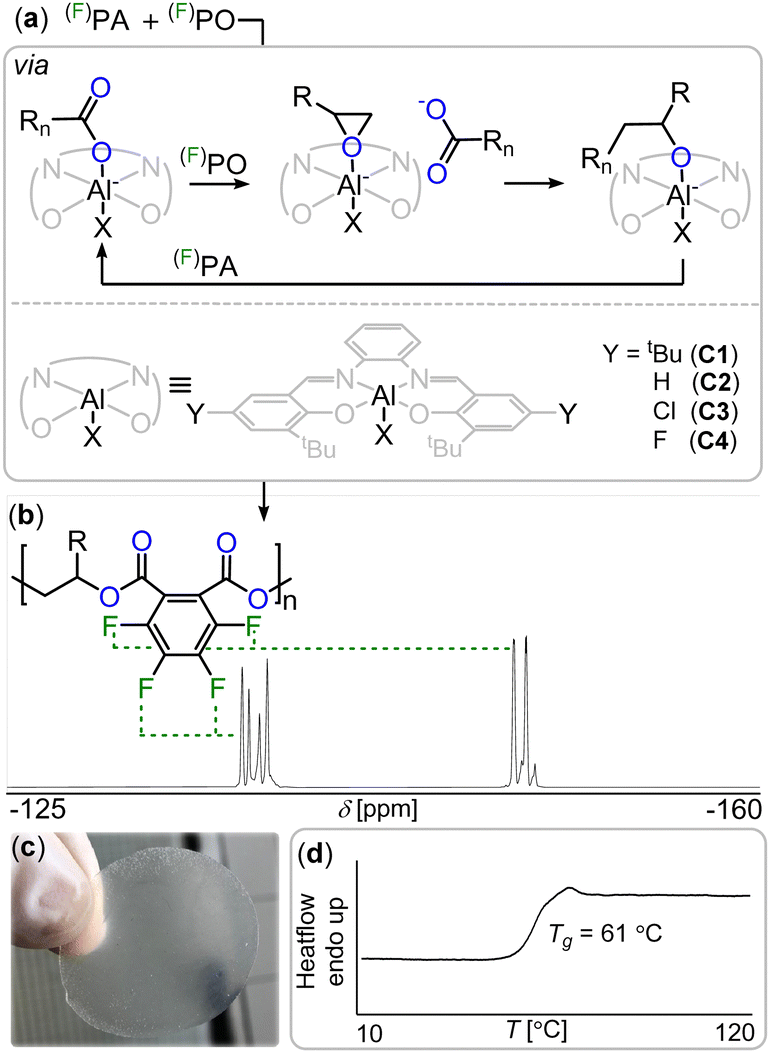 | ||
Fig. 2 (a) Intermediate speciation during (F)PA/(F)PO ROCOP under LAl(III)Cl/PPNCl catalysis. R = CH3(PO), CF3(FPO), Rn = polymer chain, X = Cl in pre-catalyst and alkoxide RnO or carboxylate RnC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O chain ends during ROCOP. (b) 19F NMR spectrum (CDCl3) of fluorinated diester unit. (c) Photograph and (d) DSC thermogram of FPA/FPO copolymer. O)O chain ends during ROCOP. (b) 19F NMR spectrum (CDCl3) of fluorinated diester unit. (c) Photograph and (d) DSC thermogram of FPA/FPO copolymer. | ||
Unlike with conventional fluorinated polymers, the presence of an ester bond in the backbone allows for degradation via nucleophilic attack of the main chain.41 Accordingly, degradation studies of the film-processed polymers with different degrees of fluorination were carried out under basic conditions employing 5 wt% NaOH in 6:4 EtOH:H2O at 40 °C as previously optimised for semi-aromatic polyesters.42 Surprisingly the more fluorinated the polymer, the faster the degradation, meaning that films comprising of 100 mg polymer at identical dimensions took 6 h for FPA/FPO, 72 h for FPA/PO and 144 h for PA/PO to fully degrade. This is even more unexpected when considering that increasing fluorination causes the materials to be more hydrophobic. Water contact angle measurements obtained from surface coated silicon wafers reveal a contact angle of 97.4° for FPA-co-FPO, 90.5° for FPA-co-PO and 80.9° for PA-co-PO.
GPC analysis of a partially degraded film revealed identical traces to the initially prepared samples indicating a surface rather than a bulk erosion mechanism (see Fig. S61, ESI†). Scanning electron microscopy of the cross section of a partially degraded film confirmed this notion showing a rough surface and a smooth homogenous interior (see Fig. 3). In the case of the polymers from FPA featuring aromatically bound fluorine, 19F-NMR analysis of the degradation products shows a multitude of arene bound fluorine multiplet resonances between −137 and −154 ppm rather than two sets of resonances for a perfluorinated phthalate ring (see Fig. S74, ESI†). Furthermore, a sharp singlet at −122 ppm is formed, which can be assigned to sodium fluoride (NaF). This can be rationalised considering a defluorination pathway via nucleophilic aromatic substitution in which an alkoxide or hydroxide anion attacks an arylic sp2-carbon centre forming a tetrahedral sp3-carbon that thereafter eliminates inorganic fluoride.43 To investigate whether full defluorination of the aromatic backbone can be achieved, the FPA derived polymer films were subjected to a 5 wt% sodium methoxide solution in methanol and left to stir at 110 °C for 8 h (see Fig. 4). Again, a de-symmetrised aromatic region in the 19F spectrum was observed, which however eventually disappeared completely so that all arene bound fluorine was transformed into NaF. Sodium fluoride precipitates from the degradation mixture together with some defluorinated carboxylate salts allowing for its easy separation by centrifugation. We hypothesized that as degradation of our materials generates inorganic fluoride salts this precipitate could be employed to generate HF as with CaF2 (Fig. 1). This would effectively recover the arene bound fluoride of the copolymers in order to allow it to be reintroduced into our current fluorine economy. Acidification of the precipitate with H2SO4 leads to visible gas formation which we attributed to HF. To simplify spectroscopic analysis, we isolated the gas by condensation into d5-pyridine. 19F NMR analysis shows appearance of a clean singlet at −172 ppm which indeed corresponds to so-called Olah's reagent, the HF adduct of pyridine (see Fig. 4).44 The supernatant of the polymer degradation with sodium methoxide in methanol contains pure propane-diol in the case of the FPA-co-PO degradants or the trifluoromethyl derivative in the case of FPA-co-FPO degradants and these diols can be isolated by distillation. To understand the difference in methanolysis rate that fluorination brings about as well as the defluorination pathway we turned to DFT modelling (see Notes S11, ESI†). A simplified ortho-dimethyl phthalate model system featuring either a perfluorinated or non-fluorinated aromatic group which is attacked by a methoxide anion (which NaOMe provides) was investigated as this process can ultimately lead to chain scission and hence degradation of the material (see Fig. S58, ESI†).
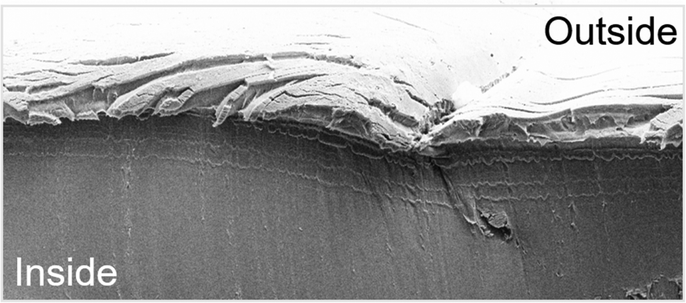 | ||
| Fig. 3 Scanning electron microscopy image of the cross-section of PA/PO film after partial degradation. | ||
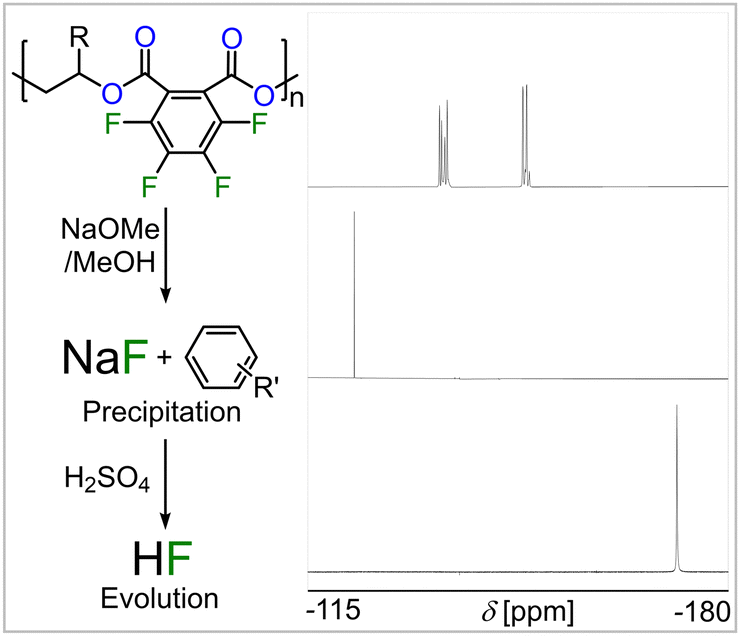 | ||
| Fig. 4 Stacked 19F NMR of (top) FPA copolymer (in CDCl3), (middle) NaF (in D2O) generated by methanolysis and HF (in d5-pyridine) by H2SO4 treatment. | ||
For the non-fluorinated case, carbonyl attack leading to a tetrahedral intermediate was found to be endergonic by 5.9 kJ mol−1 after a reaction barrier of 41.9 kJ mol−1. Contrarily, in the fluorinated case formation of the corresponding intermediate is exergonic releasing –19.2 kJ mol−1 while having to overcome a barrier of 31.7 kJ mol−1. This shows that nucleophilic attack of the FPA derived polymer is likely both thermodynamically and kinetically more favourable than for their non-fluorinated PA derived counterparts. As degradation appears to occur via surface erosion, the polar medium only has to diffuse into the hydrophobic material to a limited extent. Hence the increase in susceptibility to nucleophilic carbonyl attack appears to be more influential than the increase in hydrophobicity fluorination brings about which helps to explain fluorination enhanced degradability. We believe that the electron withdrawing nature of the fluorine substituents are responsible for this rendering the carbonyl carbon more electron deficient and hence more readily attacked by electron rich hydroxide and alkoxide nucleophiles. With regards to defluorination, the initial formation of a highly de-symmetrised spectrum indicates different co-occurring pathways which in the simplest case can be attributed to nucleophilic attack of alkoxide in either meta or para position of the ethyl-ester substituents. Here the intermediate from meta-attack forms exergonically releasing −40.3 kJ mol−1 after a 54.8 kJ mol−1 barrier while the intermediate from para-attack forms exergonically releasing −63.0 kJ mol−1 after a 47.6 kJ mol−1 barrier showing that the latter case is both kinetically and thermodynamically favoured over the former. Moreover, the activation barriers of these processes indicate that chain scission via attack of the carbonyl carbon likely occurs prior to defluorination, although in order to make a definitive statement consideration of discrete solvation as well as coordination of the counteraction would need to be considered. Nevertheless, the data indicates that the more exergonic attack at the arene ring could also positively contribute to the fluorinated materials degradability particularly at increased temperature.
In conclusion we present the ring opening copolymerisation of tetrafluorophthalic anhydride to give access to fluorinated polyesters as well as their block polymers. Although rendering the material more hydrophobic, fluorination was found to accelerate degradation via surface erosion by up to a factor of 20 which could be partially attributed to the increased susceptibility to nucleophilic attack of the carbonyl group. Arene bound fluorine can be recovered via nucleophilic aromatic substitution as inorganic fluorides that evolve HF upon acidification. Following our results the design of future fluorinated polymers should feature in-built degradation and recycling options to approach a more circular fluorine economy.
This project is part of the CRC 1349 (DFG). Computing time was made available by the High-Performance Computing Centre at the ZEDAT. We also thank the Core Facility BioSupraMol.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Améduri, Molecules, 2023, 28, 7564 CrossRef PubMed.
- J. Wiegmann, S. Beuermann and A. P. Weber, Chem. Ing. Tech., 2021, 93, 1300–1306 CrossRef CAS.
- B. Ameduri, Chem. – Eur. J., 2018, 24, 18830–18841 CrossRef CAS.
- S. Shabanian, S. K. Lahiri, M. Soltani and K. Golovin, Mater. Today Chem., 2023, 34, 101786 CrossRef CAS.
- M. Yu, W. Lyu, Y. Liao and M. Zhu, Adv. Fiber Mater., 2023, 5, 543–553 CrossRef CAS.
- S. Y. Wee and A. Z. Aris, NPJ Clean Water, 2023, 6, 57 CrossRef CAS.
- A. F. Peritore, E. Gugliandolo, S. Cuzzocrea, R. Crupi and D. Britti, Int. J. Mol. Sci., 2023, 24, 11707 CrossRef CAS PubMed.
- T. Stoiber, S. Evans and O. V. Naidenko, Chemosphere, 2020, 260, 127659 CrossRef CAS PubMed.
- E. Hepburn, C. Madden, D. Szabo, T. L. Coggan, B. Clarke and M. Currell, Environ. Pollut., 2019, 248, 101–113 CrossRef CAS PubMed.
- H. Brake, A. Langfeldt, J. Kaneene and M. Wilkins, J. Am. Vet. Med. Assoc., 2023, 261, 952–958 CAS.
- J. Garnett, C. Halsall, A. Vader, H. Joerss, R. Ebinghaus, A. Leeson and P. Wynn, Environ. Sci. Technol., 2021, 55, 11049–11059 CrossRef CAS.
- D. Sathe, J. Zhou, H. Chen, B. R. Schrage, S. Yoon, Z. Wang, C. J. Ziegler and J. Wang, Polym. Chem., 2022, 13, 2608–2614 RSC.
- B. Améduri and H. Hori, Chem. Soc. Rev., 2023, 52, 4208–4247 RSC.
- C. F. Z. Lacson, M.-C. Lu and Y.-H. Huang, Chemosphere, 2020, 239, 124662 CrossRef CAS PubMed.
- T. S. Hayes, M. M. Miller, G. J. Orris and N. M. Piatak, Fluorine, Report 1411339916, US Geological Survey, 2017.
- M. Morita, G. Granata and C. Tokoro, Mater. Trans., 2018, 59, 290–296 CrossRef CAS.
- E. Iacovidou, J. Millward-Hopkins, J. Busch, P. Purnell, C. A. Velis, J. N. Hahladakis, O. Zwirner and A. Brown, J. Clean. Prod., 2017, 168, 1279–1288 CrossRef.
- T. M. McGuire, A. Buchard and C. Williams, J. Am. Chem. Soc., 2023, 145, 19840–19848 CrossRef CAS PubMed.
- Z. Zhou, A. M. LaPointe, T. D. Shaffer and G. W. Coates, Nat. Chem., 2023, 15, 856–861 CrossRef CAS PubMed.
- X. Lu, X. Zhang, C. Zhang and X. Zhang, Adv. Sci., 2024, 11, 2306072 CrossRef CAS PubMed.
- K. A. Andrea, M. D. Wheeler and F. M. Kerton, Chem. Commun., 2021, 57, 7320–7322 RSC.
- J. Xu and N. Hadjichristidis, Prog. Polym. Sci., 2023, 101656 CrossRef CAS.
- K. M. Osten and P. Mehrkhodavandi, Acc. Chem. Res., 2017, 50, 2861–2869 CrossRef CAS PubMed.
- J. Bruckmoser, S. Remke and B. Rieger, ACS Macro Lett., 2022, 11, 1162–1166 CrossRef CAS PubMed.
- J. J. Reisinger and M. A. Hillmyer, Prog. Polym. Sci., 2002, 27, 971–1005 CrossRef CAS.
- S. Lingier, R. Szpera, B. Goderis, B. Linclau and F. E. Du Prez, Polymer, 2019, 164, 134–141 CrossRef CAS.
- R. C. Reis-Nunes, E. Riande, N. C. Chavez and J. Guzmán, Macromolecules, 1996, 29, 7989–7994 CrossRef.
- G. H. He, B. H. Ren, S. Wang, Y. Liu and X. B. Lu, Angew. Chem., 2023, 135, e202304943 CrossRef.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2021, 60, 13372–13379 CrossRef CAS PubMed.
- R. Xie, Y. Y. Zhang, G. W. Yang, X. F. Zhu, B. Li and G. P. Wu, Angew. Chem., 2021, 133, 19402–19410 CrossRef.
- W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed.
- A. J. Plajer and C. K. Williams, ACS Catal., 2021, 11, 14819–14828 CrossRef CAS.
- Y. Manjarrez, M. D. C. L. Cheng-Tan and M. E. Fieser, Inorg. Chem., 2022, 61, 7088–7094 CrossRef CAS PubMed.
- F. D. Monica and A. W. Kleij, ACS Sustainable Chem. Eng., 2021, 9, 2619–2625 CrossRef CAS.
- F. Fiorentini, W. T. Diment, A. C. Deacy, R. W. Kerr, S. Faulkner and C. K. Williams, Nat. Commun., 2023, 14, 4783 CrossRef CAS PubMed.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS PubMed.
- B. Cappella and G. Dietler, Surf. Sci. Rep., 1999, 34, 1–104 CrossRef CAS.
- M. Dudziak, I. Topolniak, D. Silbernagl, K. Altmann and H. Sturm, Nanomater, 2021, 11, 3285 CrossRef CAS PubMed.
- D. Silbernagl, N. C. Murillo, A. M. Elert and H. Sturm, Beilstein J. Nanotechnol., 2021, 12, 58–71 CrossRef CAS PubMed.
- R. W. Kerr and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 6882–6893 CrossRef CAS PubMed.
- S. Ügdüler, K. V. Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. D. Meester, Green Chem., 2020, 22, 5376–5394 RSC.
- S. Mallick, P. Xu, E. U. Würthwein and A. Studer, Angew. Chem., 2019, 131, 289–293 CrossRef.
- G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872–3881 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02513j |
| This journal is © The Royal Society of Chemistry 2024 |