
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of macrocyclic thiolactone peptides via photochemical intramolecular radical acyl thiol–ene ligation†
Alby
Benny
 and
Eoin M.
Scanlan
*
and
Eoin M.
Scanlan
*
Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse Street, Dublin 2, Ireland. E-mail: eoin.scanlan@tcd.ie
First published on 5th July 2024
Abstract
A photochemical acyl thiol–ene reaction can be used to rapidly cyclise fully unprotected peptides bearing both a thioacid and alkene to form peptide thiolactones. This strategy represents the first reported synthesis of peptide thiolactones under radical-mediated conditions.
Cyclic peptides have gained considerable interest in recent years both as drugs and as chemical probes due to their conformational rigidity, which results in improved metabolic stability, membrane permeability, selectivity and binding affinity compared to the linear counterparts.1,2 The majority of clinically approved cyclic peptide drugs to date have been derived from naturally occurring cyclic peptides such as hormones or antimicrobial agents.3,4 However, the de novo development of cyclic peptides is becoming increasingly common with the aid of computational design and high-throughput screening of synthetic cyclic peptide libraries.5
Common peptide cyclisation strategies include macrolactamisation, macrolactonisation, disulfide formation and ring closing metathesis.6,7 In addition, a variety of two-component peptide stapling methods have been developed whereby a bifunctional linker is used to crosslink two reactive groups on the linear peptide.8 Click reactions such as the copper-catalysed azide–alkyne cycloadditon (CuAAC) and thiol–ene reactions have emerged as rapid, highly efficient methods for the synthesis of cyclic peptides.9,10 The photochemical thiol–ene reaction involves the radical-mediated anti-Markovnikov addition of a thiol onto an alkene to form a thioether linkage.11 The fast kinetics, high yields, mild reaction conditions, compatibility with aqueous conditions, and chemoselectivity of the thiol–ene reaction has prompted its widespread application for peptide bioconjugation and macrocyclisation.10,12 The absence of any metal catalysts is a notable advantage of the thiol–ene reaction compared to the CuAAC reaction, as Cu(I) can be cytotoxic and complete removal of the metal can be labourious.13
Recently, we reported the radical-mediated acyl thiol–ene reaction to synthesise a variety of thioesters and small-molecule thiolactones through direct addition of thioacids onto alkenes.14–16 Thiolactones are found in several bioactive natural depsipeptides such as thiocoraline and verrucosamide.17,18 Another class of macrocyclic thiolactones known as autoinducing peptides (AIPs) are found in Gram positive bacteria such as Staphylococcus aureus and have a key role in the regulation of the quorum sensing system and the production of virulence factors.19–21 Although thioesters are unstable under basic conditions, analogues of AIPs have been successfully investigated as modulators of the quorum sensing system to attenuate and treat S. aureus infections.22–24
Current methods for the chemical synthesis of macrocyclic thiolactone peptides have been reviewed by Gordon.25 In summary, early approaches utilising coupling reagents to form the thiolactone bond between a cysteine (Cys) residue and the C-terminal carboxylic acid require fully protected peptides and long reaction times (Fig. 1a). More recent approaches whereby Cys displaces a latent thioester or activated leaving group (Dbz, MeDbz) to form the thiolactone still require reaction times of 2 h at elevated temperatures for good conversion (Fig. 1b). In this study, we describe a rapid photochemical acyl thiol–ene cyclisation strategy for fully unprotected peptides bearing a thioacid and alkene to furnish macrocyclic thiolactone peptides (Fig. 1c).
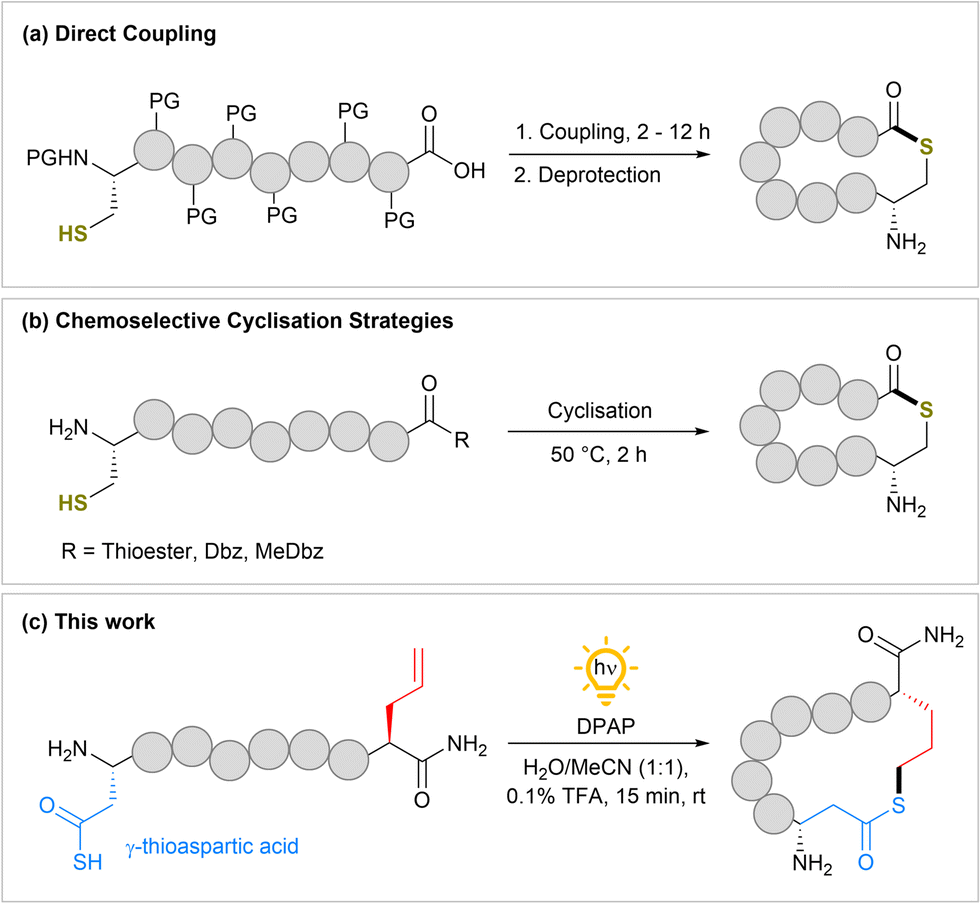 | ||
| Fig. 1 Synthesis of peptide thiolactones. (a) Direct coupling of protected peptides to form thiolactones. (b) Chemoselective thiolactone formation via displacement of activated C-terminal groups. (c) This work; photochemical synthesis of peptide thiolactones via intramolecular radical-mediated acyl thiol–ene. PG = Protecting group. | ||
Previous work by Shimuzu et al. demonstrated that C-terminal peptide thioacid radicals undergo dethiocarboxylation via carbonyl sulfide (COS) elimination to form N-alkylamides.26 In order to avoid this side reaction during acyl thiol–ene macrocyclisation, we utilised γ-thioaspartic acid bearing the required thioacid on the side chain of aspartic acid (Asp) as dethiocarboxylation should be unfavourable due to the requisite formation of a primary alkyl radical intermediate.27 We envisaged introducing the γ-thioaspartic acid into the linear peptide as the protected S-triphenylmethyl (STrt) thioester through standard Fmoc solid phase peptide synthesis (SPPS), with subsequent deprotection to release the free thioacid during resin cleavage upon treatment with trifluoroacetic acid (TFA). To this end, the Asp(γ-STrt) derivative 1 was synthesised to serve as the building block for SPPS (Scheme 1). Previous work has shown that Asp(γ-STrt) thioesters are susceptible to aspartimide formation during Fmoc deprotection in SPPS; therefore the N-terminus was protected with the acid-labile tert-butyloxycarbonyl (Boc) group which is removed during resin cleavage.28,29 Consequently, compound 1 must be incorporated in SPPS as the final amino acid at the N-terminal of the peptide sequence.
 | ||
| Scheme 1 Synthesis of Boc-Asp(STrt)-OTMS 1. | ||
To commence our study, the allyl (All) ester Boc-Asp-OAll (3) was synthesised from commercially available Boc-Asp(OtBu)-OH (2) following the procedure reported by Kapurniotu and Taylor (Scheme 1).30 Steglich thioesterification of the side chain carboxylic acid with triphenylmethanethiol (TrtSH) in dichloromethane (DCM) formed the Asp(STrt) thioester 4 in an isolated yield of 63%. Selective removal of the C-terminal allyl group via Pd-mediated deallylation furnished the carboxylic acid derivative 5 in 90% yield. The C-terminal carboxylic acid of side-chain trityl thioesters can undergo base-catalysed cyclic anhydride formation during activation via elimination of triphenylmethanethiol, making the C-terminal carboxylic acid 5 unsuitable for use in SPPS.28,29 However, Joseph et al. have demonstrated that this undesirable side-reaction can be suppressed through formation of the corresponding C-terminal trimethylsilyl (TMS) ester which acts as a carboxylic acid surrogate while remaining sufficiently reactive for amide coupling with N,N′-diisoproylcarbodiimide (DIC).29 Thus, compound 5 was reacted with trimethylsilyl chloride (TMSCl) for 30 min to form the required TMS ester 1 in quantitative yield. Overall, Boc-Asp(STrt)-OTMS (1) was prepared from compound 2 in a yield of 51% over 6 steps, requiring only two chromatographic purification steps. The compound is stable for several months at 0 °C if stored under a layer of inert gas and care is taken to exclude moisture.
Having prepared the requisite building block, we next set out to synthesise linear peptides bearing both an alkene and thioacid suitable for acyl thiol–ene cyclisation. The terminal alkene allylglycine (Agl) was selected for this study because it is commercially available and is stable to standard Fmoc SPPS conditions. The terminal alkene also ensures that only a single isomer of the cyclic peptide is formed upon acyl thiol–ene cyclisation because anti-Markovnikov addition of the thioacid radical to the alkene is highly favoured.16 Fmoc SPPS using Rink amide resin was used to synthesise the model linear peptide H-Asp(SH)-Ala-Ala-Agl-NH26 (Scheme 2). HATU/DIPEA in DMF was used for all amino acid couplings, except for Boc-Asp(STrt)-OTMS 1, which was coupled using DIC/Oxyma Pure/DIPEA in dry DCM. Following cleavage, the fully unprotected linear peptide 6 was obtained in a yield of 83%. The peptide was obtained in sufficient purity for use in macrocyclisation studies without the requirement for further HPLC purification.
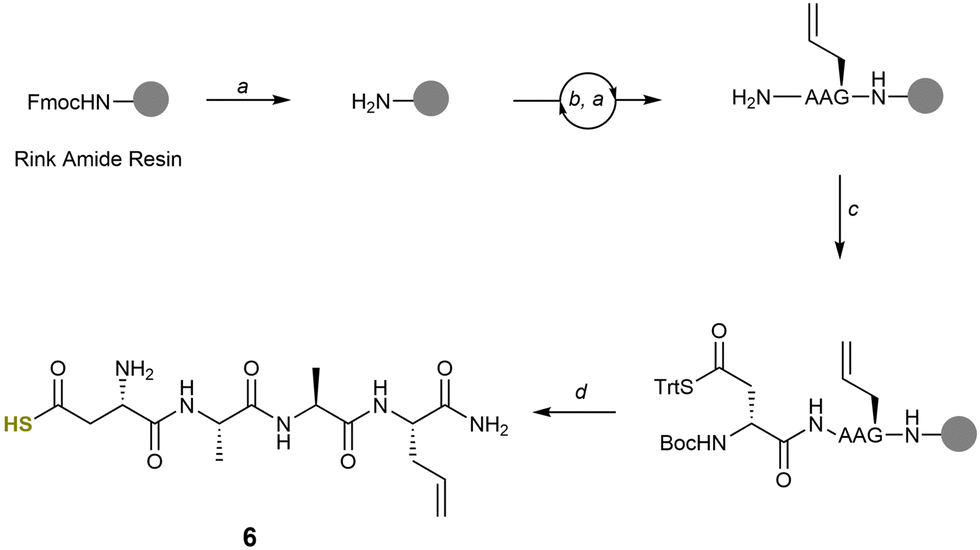 | ||
Scheme 2 Solid phase peptide synthesis of 6. (a) 20% Piperidine/DMF, 10 min × 2. (b) Fmoc-AA-OH (4 eq.), HATU (3.9 eq.), DIPEA (8 eq.), DMF (0.2 M), 45 min. (c) Boc-Asp(STrt)-OTMS 1 (3 eq.), DIC (3 eq.), Oxyma Pure (3 eq.), DIPEA (3 eq.), Dry DCM (0.2 M), 45 min × 2 (d) TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM:TES:H2O (90:5:2.5:2.5), 2 h. :DCM:TES:H2O (90:5:2.5:2.5), 2 h. | ||
The linear peptide 6 was dissolved at 5 mM concentration in a solvent mix of H2O/MeCN (1:1) containing 0.1% TFA and one eq. of the photoinitiator, 2,2-dimethoxy-2-phenylaceto- phenone (DPAP), and irradiated with UV light (354 nm) for 15 min. Gratifyingly, a conversion of 90% to the thiolactone 7 was observed by analytical HPLC (Fig. 2b) and the product was obtained in 54% yield following semi-preparative HPLC purification. Since the acyl thiol–ene macrothiolactonisation is an intramolecular hydrothioacylation reaction, the mass of the linear and cyclised peptides are identical. Therefore, along with the shift to lower retention time observed in the analytical HPLC trace (Fig. 2b), NMR spectroscopy was used to confirm formation of the peptide thiolactone 7. The HMBC spectrum of 7 provided in Fig. 2c shows unambiguous evidence for the formation of the thiolactone linkage. The HMBC spectrum shows correlation between the thiolactone carbonyl at position #2 (13C δ 194.8 ppm) and the diastereotopic methylene protons at position #3 (1H δ 3.13–3.07 and δ 2.59–2.51 ppm).
 | ||
| Fig. 2 (a) ATE macrocyclisation of 6. Reaction conditions: peptide 6 (5 mM), DPAP (5 mM) in H2O/MeCN (1:1) containing 0.1% TFA, UV light (354 nm), rt, 15 min. (b) (i) HPLC trace of linear peptide 6 (5–95% over 10 min, 220 nm) (ii) HPLC trace of reaction mixture at 15 min (5–95% over 10 min, 220 nm). *DPAP by-products. (c) HMBC spectrum of 7 showing correlation (blue arrows) across the thiolactone bond. | ||
The acyl thiol–ene cyclisation of 6 without the addition of 0.1% TFA in the solvent system resulted in a significantly reduced conversion of 35% to the thiolactone 7 (Fig. S2 in ESI†). As previously observed for the thiol–ene reaction, the addition of TFA in the solvent mixture is necessary to ensure that the thioacid remains protonated for efficient hydrogen atom transfer (HAT) to form the requisite thiyl radical to initiate the reaction.31 Screening of reaction time showed a moderate conversion of 59% at just 1 min irradiation time, and up to 80% at 5 min (Fig. S3 and S4 in ESI†). The reaction was conducted with 25 mM concentration of the linear peptide and a conversion of 82% was observed without significant intermolecular dimerisation or polymerisation reactions (Fig. S5 in ESI†).
To investigate the scope of the acyl thiol–ene mediated macrothiolactonisation reaction, a series of structurally diverse tetramer and pentamer linear peptides S2–S4 were synthesised by SPPS (structures provided in ESI†). After cyclisation using the standard conditions, thiolactone peptides 8–10 were obtained in moderate to good isolated yields (Fig. 3). In the case of 11, no cyclisation was observed under the standard conditions. We theorised that intramolecular hydrogen bonding of the thioacid to the adjacent serine residue was preventing proton abstraction by the photoinitiator. Thus, the reaction was repeated with the addition of the chaotropic agent, 6 M guanidine hydrochloride, in the solvent mixture. Gratifyingly, under these modified conditions, full consumption of the linear peptide was observed by HPLC, and thiolactone 11 was isolated in a yield of 61%. Importantly, side-chain functional groups including phenols (8), amines (9), carboxylic acids (10) and alcohols (11) were all compatible with the acyl thiol–ene macrocyclisation protocol described. Furthermore, the sequence of thiolactone peptides 10 and 11 were based on the structure of the autoinducing peptides AIP-I and AIP-II, highlighting the utility of the reaction for the synthesis of biologically relevant peptides. Linear peptides S5 and S6 were also synthesised and cyclised to form thiolactones S7 and S8 (structures provided in ESI†). However, S7 and S8 were isolated with approximately 20% of a coeluting impurity corresponding to truncated or hydrolysed linear thioacid peptide carried through from the SPPS synthesis. Nonetheless, these peptides are still of sufficient purity (>75%) for initial, high-throughput drug screening studies.
 | ||
| Fig. 3 Scope of the acyl thiol–ene cyclisation. Isolated yields after semi-preparative HPLC purification. Conditions unless otherwise stated for cyclisation: linear peptide (5 mM), DPAP (1 eq.) in H2O/MeCN (1:1) + 0.1% TFA, UV light (354 nm), rt, 15 min. a6 M guanidine hydrochloride added to solvent mixture. | ||
In summary, we report the use of the photochemical acyl thiol–ene reaction for the synthesis of peptide thiolactones starting from unprotected linear peptides bearing γ-thioaspartic acid and allylglycine residues. The cyclisation reaction is rapid (15 min) and chemoselective and represents the first reported synthesis of peptide thiolactones under photochemical, radical-mediated conditions. The rapid nature of the acyl thiol–ene thiolactonisation reaction would be amenable to the synthesis of peptide libraries, particularly with the development of on-resin cyclisation protocols. Further studies towards the application of this methodology for the preparation of libraries of novel thiolactone peptides suitable for high-throughput screening are currently ongoing in our labs.
Conceptualisation, E. M. S.; methodology, A. B.; writing – original draft preparation, A. B.; writing – review and editing, E. M. S.; supervision, E. M. S.; funding acquisition, E. M. S.
This work was funded by the Irish Research Council (Grant GOIPG/2020/813) and Science Foundation Ireland (19/FFP/6667). We are grateful to Dr John O’Brien and Dr Manuel Ruether for assistance with NMR spectroscopy and Dr Gary Hessman for assistance with mass spectrometry.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- D. S. Nielsen, R.-J. Lohman, H. N. Hoang, T. A. Hill, A. Jones, A. J. Lucke and D. P. Fairlie, ChemBioChem, 2015, 16, 2289–2293 CrossRef CAS PubMed.
- L. Mendive-Tapia, J. Wang and M. Vendrell, Pept. Sci., 2021, 113, e24181 CrossRef CAS.
- X. Ji, A. L. Nielsen and C. Heinis, Angew. Chem., Int. Ed., 2024, 63, e202308251 CrossRef CAS PubMed.
- H. Zhang and S. Chen, RSC Chem. Biol., 2022, 3, 18–31 RSC.
- M. L. Merz, S. Habeshian, B. Li, J.-A. G. L. David, A. L. Nielsen, X. Ji, K. Il Khwildy, M. M. Duany Benitez, P. Phothirath and C. Heinis, Nat. Chem. Biol., 2023, 20, 634 Search PubMed.
- C. Bechtler and C. Lamers, RSC Med. Chem., 2021, 12, 1325–1351 RSC.
- A. R. Paquette and C. N. Boddy, Org. Biomol. Chem., 2023, 21, 8043–8053 RSC.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- R. A. Turner, A. G. Oliver and R. S. Lokey, Org. Lett., 2007, 9, 5011–5014 CrossRef CAS PubMed.
- M. D. Nolan and E. M. Scanlan, Front. Chem., 2020, 8, 583272 CrossRef CAS PubMed.
- A. A. Aimetti, R. K. Shoemaker, C.-C. Lin and K. S. Anseth, Chem. Commun., 2010, 46, 4061–4063 RSC.
- M. Ahangarpour, I. Kavianinia, P. W. R. Harris and M. A. Brimble, Chem. Soc. Rev., 2021, 50, 898–944 RSC.
- L. Li and Z. Zhang, Molecules, 2016, 21, 1393 CrossRef PubMed.
- R. O. McCourt, F. Dénès, G. Sanchez-Sanz and E. M. Scanlan, Org. Lett., 2018, 20, 2948–2951 CrossRef CAS PubMed.
- R. O. McCourt and E. M. Scanlan, Org. Lett., 2019, 21, 3460–3464 CrossRef CAS PubMed.
- J. T. McLean, P. Milbeo, D. M. Lynch, L. McSweeney and E. M. Scanlan, Eur. J. Org. Chem., 2021, 4148–4160 CrossRef CAS.
- V. Nair, M. C. Kim, J. A. Golen, A. L. Rheingold, G. A. Castro, P. R. Jensen and W. Fenical, Mar. Drugs, 2020, 18, 549 CrossRef CAS PubMed.
- F. Romero, F. Espliego, J. Pérez Bas, T. García de Quesada, D. Grávalos, F. De la Calle and J. Luis Fernández-Puentes, J. Antibiot. Res, 1997, 47, 734–737 CrossRef PubMed.
- B. Wang and Tom W. Muir, Cell Chem. Biol., 2016, 23, 214–224 CrossRef CAS PubMed.
- Y. Tal-Gan, M. Ivancic, G. Cornilescu, C. C. Cornilescu and H. E. Blackwell, J. Am. Chem. Soc., 2013, 135, 18436–18444 CrossRef CAS PubMed.
- B. H. Gless, M. S. Bojer, P. Peng, M. Baldry, H. Ingmer and C. A. Olsen, Nat. Chem., 2019, 11, 463–469 CrossRef CAS PubMed.
- J. K. Vasquez and H. E. Blackwell, ACS Infect. Dis., 2019, 5, 484–492 CrossRef CAS PubMed.
- C. P. Gordon, S. D. Olson, J. L. Lister, J. S. Kavanaugh and A. R. Horswill, J. Med. Chem., 2016, 59, 8879–8888 CrossRef CAS PubMed.
- K. H. J. West, C. G. Gahan, P. R. Kierski, D. F. Calderon, K. Zhao, C. J. Czuprynski, J. F. McAnulty, D. M. Lynn and H. E. Blackwell, Angew. Chem., Int. Ed., 2022, 61, e202201798 CrossRef CAS PubMed.
- C. P. Gordon, Org. Biomol. Chem., 2020, 18, 379–390 RSC.
- T. Shimizu, R. Miyajima, N. Naruse, K. Yamaoka, K. Aihara, A. Shigenaga and A. Otaka, Chem. Pharm. Bull., 2016, 64, 375–378 CrossRef CAS PubMed.
- A. Benny, L. Di Simo, L. Guazzelli and E. M. Scanlan, Molecules, 2024, 29, 1465 CrossRef CAS PubMed.
- M. J. Schöwe, O. Keiper, C. Unverzagt and V. Wittmann, Chem. – Eur. J., 2019, 25, 15759–15764 CrossRef PubMed.
- R. Joseph, M. Morales Padilla and P. Garner, Tetrahedron Lett., 2015, 56, 4302–4304 CrossRef CAS.
- A. Kapurniotu and J. W. Taylor, Tetrahedron Lett., 1993, 34, 7031–7034 CrossRef CAS.
- M. D. Nolan, C. Shine, E. M. Scanlan and R. Petracca, Org. Biomol. Chem., 2022, 20, 8192–8196 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: synthetic details, characterisation, HPLC traces and NMR spectra. See DOI: https://doi.org/10.1039/d4cc02442g |
| This journal is © The Royal Society of Chemistry 2024 |