
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light enhanced catalytic activity of Aun subnanoclusters: the importance of d–sp interband transitions†
Alba
Sorroche
 a,
Irene
del-Campo
a,
Alberto
Casu
b,
Andrea
Falqui
b,
Miguel
Monge
*a and
José M.
López-de-Luzuriaga
*a
a,
Irene
del-Campo
a,
Alberto
Casu
b,
Andrea
Falqui
b,
Miguel
Monge
*a and
José M.
López-de-Luzuriaga
*a
aDepartamento de Química, Instituto de Investigación en Química (IQUR), Universidad de La Rioja, Complejo Científico-Tecnológico, 26006, Logroño, Spain. E-mail: miguel.monge@unirioja.es; josemaria.lopez@unirioja.es
bCIMAINA and Departimento di Fisica, Università degli Studi di Milano, Via Celoria 16, 20133, Milano, Italy
First published on 3rd July 2024
Abstract
Polymers can serve as an effective matrix to stabilize gold nanoparticles. These materials offer a continuous light-activated supply of subnanoclusters, which are composed of a few atoms. We report an efficient approach to enhance the catalytic activity of gold subnanoclusters by in situ feeding of these species through the generation of hot carriers via 5d–6s6p interband transitions on PEG-stabilized Au nanoparticles.
Gold subnanoclusters (Au SNCs) constitute a special class of catalysts showing excellent activity in several organic transformations in solution, including C–C, C–Br or C–O bond formation.1 These catalytic species consist of ligand-free clusters of 3–8 gold atoms, exhibiting molecular-type behaviour, with an electronic structure of defined molecular orbitals, in contrast to gold nanoparticles (Au NPs), which display an electronic band structure. The main drawback of Au SNCs is their transient existence, showing a large tendency to aggregate; however, they can be temporarily stabilized by a cooperative network of dispersive Au⋯C–H and/or Au⋯π interactions, which improves their catalytic performance.2 Another possibility for overcoming the short transient existence of Au SNCs during a catalytic process would be a continuous and sustained feeding of Au SNCs in the reaction media.
There are several approaches for the in situ synthesis in solution of ligand-free Au SNCs, with the main ones being the electrochemical etching of gold plates3–5 and the reduction of gold salts.6,7 Au SNCs can also be encapsulated within polymer matrices such as hyperbranched polyethyleneimine (PEI), polyamidoamine (PAMAM) dendrimers or ethylene vinyl alcohol copolymer (EVOH), or biomolecules like aminoacids, peptides, proteins or DNA.3,5,8,9 In some of the previous cases, the gold source is Au NPs and the addition of the polymer (PEI) or biomolecules produce Au SNCs through the etching of the Au NP surface. Indeed, another possible approach for the synthesis of catalytically active Au SNCs is the chemical etching of the surface of colloidal Au NPs by adding an acid (HCl) or etching the surface of Au NPs supported on solids like charcoal, TiO2, ZnO or Al2O3 by adding iodide.9
The use of plasmonic gold nanoparticles (Au NPs) as directing agents for chemical reactions in photocatalytic processes is an area of continuous expansion.10–16 The strong shape-dependent visible/NIR light absorption ability through the excitation of the localized surface plasmon resonance (LSPR), which produces hot carriers or photothermal heating (the latter specially on anisotropic gold nanostructures), improves the (photo)catalytic performance.17,18 However, the plasmon-induced hot-carriers display extremely short lifetimes (ca. 100 fs) before leading to electron–hole recombination, too short for being involved in chemical transformations unless this recombination is somehow precluded, for instance, through plasmonic nanoparticle–semiconductor heterojunctions.19 In this regard, recent studies have proposed that in addition to the LSPR optical regime, the interband d–sp transitions also led to hot carriers of longer lifetime when photons of energy above 2.4 eV (below 516 nm) excite electrons from the d-band to the sp-band. These interband transitions produce hot-deep holes with a higher reduction potential compared to the ones produced through LSPR excitation, and warm electrons. Thus, in the case of spherical Au NPs, the photocatalytic activity using broad range visible light assigned to LSPR excitation can also originate from the masked tail of the interband transition below 2.4 eV. Somorjai, Alivisatos, Toste and co-workers propose that the holes generated through interband d–sp transitions on Au NPs possess a strong π-acidic character that favours the coordination of alkynylphenols, leading to the formation of Au2–Au4 species that promote the cyclization reaction by an intramolecular nucleophilic attack.20
In this context, the catalyzed hydration of alkynes is a benchmark C–O bond formation reaction widely studied. In some cases, this reaction has been evaluated under irradiation as in the case of transition metal photocatalysts such as CuCl21 or a Rh(III) porphyrin complex under visible light,22,23 or the direct photoinduced hydration in acidic solutions.23 We have previously studied this catalytic transformation by using Au SNCs, through a thermally assisted process (reflux in methanol at 60 °C).2 At this point, we wondered whether a white LED light interband assisted process would enhance the efficiency of the hydration of alkynes by using a preformed mixture of Au NPs and Au SNCs in a polymeric polyethyleneglycol (PEG) matrix. Indeed, a continuous feeding of Au SNCs to the reaction medium formed upon interband excitation of Au NPs would compensate the transient existence of subnanoclusters species, increasing their concentration and leading to the improvement of the catalytic efficiency.
Herein we show that the catalytic hydration of alkynes by Au SNCs can be enhanced by a 3-fold increase in the reaction rate, through the visible LED light-induced irradiation of PEG-stabilized Au NPs in solution, which generates interband hot carriers leading to the in situ formation of Au SNCs (see Scheme 1). We show through TDDFT calculations that highly favoured excited states lead to a clear electron–hole separation process during the d–sp interband transition and that optimized excited states lead to the initial detachment of Au3 SNCs from the Au13 cluster model.
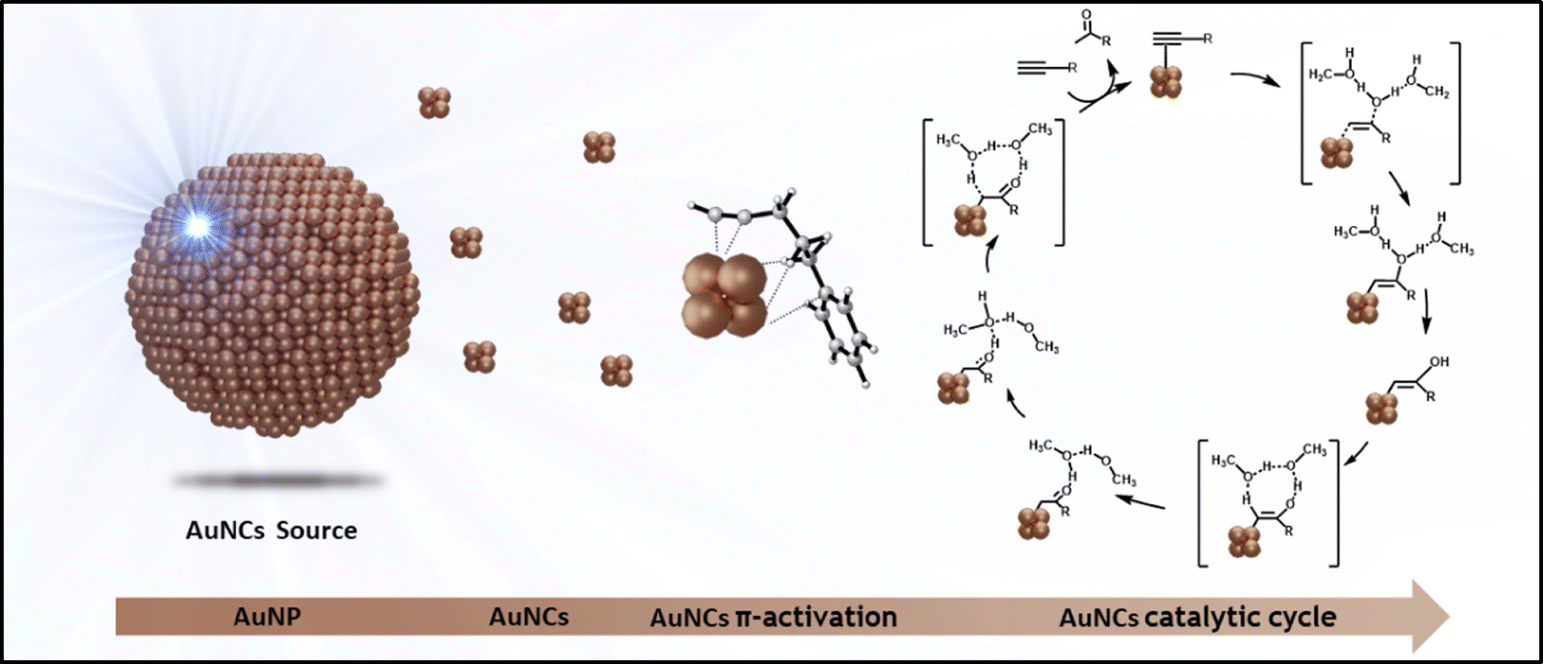 | ||
| Scheme 1 Visible light-assisted formation of Au SNCs in solution for the enhanced catalytic hydration of alkynes. | ||
Briefly, PEG-stabilized Au NPs 1 were prepared by the reduction of AuCl in refluxing MeOH, in the presence of an excess of polymer. After 1 h of reflux, the PEG-Au NP sample was obtained by solvent evaporation as a pale yellow solid.
Transmission electron microscopy (TEM) and high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) images of a PEG-Au NP sample show a very homogeneous population of small size Au seed NPs. A size analysis was performed on the images recorded in both configuration and all the results are consistent, indicating that the sample features slightly elongated Au seeds with comparable Feret and minimum Feret diameters. All size-related distributions were fitted by log-normal functions and the relevant values were calculated accordingly, with a representative aspect ratio (AR) value of 1.3 and Feret and minimum Feret diameter values of 4.8 and 3.7 nm, respectively (see Fig. 1)
 | ||
| Fig. 1 HAADF-STEM (a) and TEM (b) images of a sample of PEG-Au NPs. | ||
The presence of Au SNCs within the PEG-Au NPs was confirmed through UV-vis, fluorescence, MALDI-TOF and XPS measurements (see Fig. 2). The UV-vis spectrum of the PEG polymer shows a strong absorption in the high energy region for which a maximum is not detected above 200 nm. By contrast, upon decomposition of AuCl in the presence of PEG in MeOH, the UV-vis spectrum shows a strong absorption slightly above 200 nm, probably arising from n(O)–3s transitions in the polymer, a band at ca. 315 nm and a weak band at ca. 520 nm, which are absent in the pure PEG spectrum. Following previous studies on Au SNCs, the high energy band at 315 nm can be ascribed to the presence of subnanoclusters in solution, whereas the weak band at 520 nm can be ascribed to the LSPR mode of spherical Au NPs. The spectrum profile in the high-energy region (200–400 nm) is very similar to the one previously reported by Oliver-Messeguer et al. of Au SNCs embedded within a EVOH polymeric matrix.9 In addition, excitation of a sample of PEG-Au NPs produces a fluorescence emission at 365 nm, characteristic of Au SNCs, which is not observed when pure PEG samples are excited at similar wavelength. The MALDI-TOF spectrum in the negative mode of PEG-Au NPs also agrees with the presence of Au SNCs of Au2–Au4 composition within the polymeric matrix. The survey XPS spectrum of 1 rules out the presence of Cl atoms and, hence, of AuCl in the sample. We also recorded the high-resolution XPS spectra for the Au 4f region for PEG-Au NPs (see Fig. 2D). The doublet peak can be deconvoluted and fitted to two spin–orbit doublets, each separated in energy by ca. 3.7 eV.
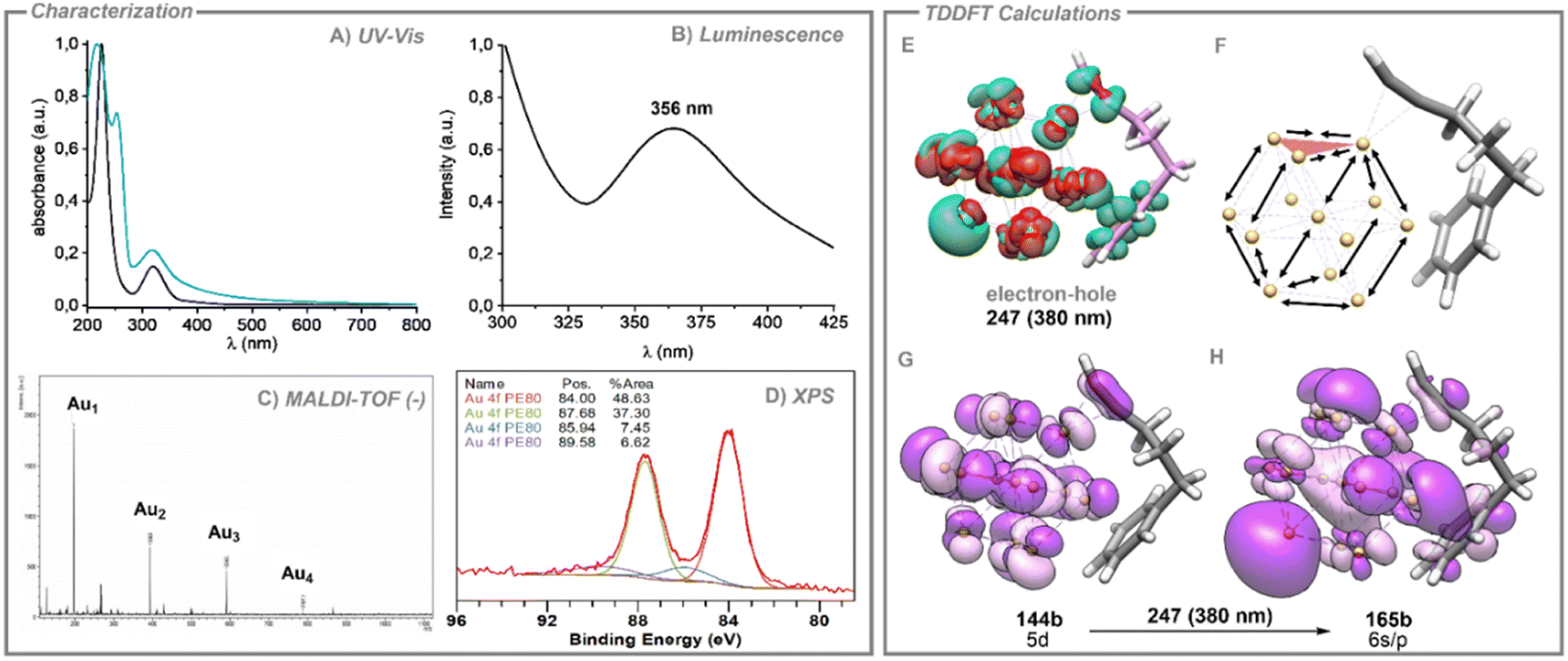 | ||
| Fig. 2 Characterization of compound Au@PEG (1a). (A) UV/vis spectra of a solution of Au@PEG (black) and Au@PEG + 5-phenyl-1-pentyne (blue). (B) Emission spectra of the compound excited at 275 nm (blue) and 250 nm (black). (C) MALDI-TOF spectra in its negative mode. (D) High-resolution XPS of the Au-4f region fitted to Au0 (low-energy doublet) and Auδ+ (high-energy doublet) composition. (E) Representation of electron–hole density (247). (F) Geometry distortion of 1a model system in 188, 232 and 247 excited states. (G) Occupied molecular orbital representation of the transition at 380 nm excitation (247). (H) Virtual molecular orbital representation of the transition at 380 nm excitation (247). | ||
The most intense doublet at 84.0 eV and 87.7 eV is assigned to metallic gold. The less intense doublet at 85.9 and 89.6 eV could be assigned to the presence of a 14% of Auδ+ charged atoms that could be attributed to the presence of Au SNCs in a similar composition to previously reported ones with the EVOH polymer.9
PEG-Au NPs 1 bearing Au SNCs were used as catalysts in the hydration of 5-phenyl-1-pentyne. The role played by visible light was studied in this catalyzed transformation by performing the same catalytic reaction in the dark and under visible LED light irradiation for 90 min. The results are depicted in Fig. S5 (ESI†) and show that under dark conditions the hydration of 5-phenyl-1-pentyne achieves lower conversions of 34% (4 mol% of added Au) of the corresponding ketone. In contrast, when the same reaction is performed under visible LED light irradiation using an irradiation power of 1147 W cm−2, the reaction proceeds with faster conversion from the beginning, leading to a high 86% of conversion after 90 min of reaction. This catalytic behaviour is reproduced using AuCl and in situ formed PEG-Au NPs 1 (ESI†).
The presence of visible light irradiation produces a positive effect in the catalytic conversion since compared to the induction time observed for the reaction carried out under dark conditions, a clearly shorter induction time is observed. Considering that in all cases Au SNCs are present from the beginning of the reaction, it seems that under dark conditions, the amount of Au SNCs formed is too low for avoiding an induction period (see Fig. S10, ESI†). However, the irradiation with visible light clearly accelerates the catalytic transformation from the beginning, leading to better conversions. It is also worth highlighting that irradiation with green LED light (532 nm) resulted in a lower catalytic conversion (15%) in contrast to employing white LED light. Moreover, the catalytic conversion of the reaction is also diminished when employing blue (31%) or UV LED light (57%) (see Table 1, entries 4 and 5), but to a lesser extent. Hence, the use of green light that exclusively excites the reaction mixture in the LSPR region yields poor catalytic results, thereby ruling out the possibility of exclusively attributing this process to a photocatalytic plasmonic process. Note that visible white LED irradiates in the 425–650 nm range with two maxima at ca. 450 and 550 nm (see Fig. S15 in the ESI†).
| Entry | Light region | Conversion (%) |
|---|---|---|
| a Reaction conditions: 4% of catalyst (Au@PEG 1) in methanol at 60 °C. Conversion determined by gas chromatography–mass spectrometry (GC-MS) at 60/90 minutes of reaction. | ||
| 1 | Dark | 20/34 |
| 2 | White | 78/86 |
| 3 | Green | 15/36 |
| 4 | Blue | 31/40 |
| 5 | UV | 57/65 |
In order to discern whether the light activation of the nanoparticles produces a photothermal or a photochemical process (near-field enhancement or hot-charge carriers), we conducted several experiments.24 For a photochemical process, the rate η (M mol−1) of the catalytic conversion is directly proportional to the rate of incident light power on the reaction. However, a photothermal process exhibits an exponential correlation with the intensity of illumination irradiance, typically fitted to an Arrhenius-type equation η = A exp(−Ea/RT) with temperature dependence. Fig. S14 (ESI†) shows that the representation of the rate of hydration (fitted as zeroth order processes) vs. irradiation power can be linearly fitted ruling out a photothermal process.
Taking into account that increasing the energy of the incident light from green to blue and to UV increases the catalytic conversion or the fact that the irradiation with broad range white light produces the best conversion results, we envisaged the possibility of producing hot-charge carriers through d–sp interband transitions upon irradiation, which could be responsible for the increased formation of catalytically active Au SNCs. To verify the occurrence of these d–sp interband transitions, we have performed DFT and TDDFT calculations on a Au13-5-phenyl-1-pentyne model system (1a). This type of model would not provide information about plasmonic absorptions because of its small size but, considering that plasmonic effects would be neglected here, we use this model system to prove the occurrence of d–sp interband transitions upon irradiation and cluster excitation. The optimized model in the S0 ground state showed the Au⋯π activation of the alkyne together with stabilized Au⋯H–C and Au⋯π(phenyl) interactions.
We computed the absorption spectrum of model 1a, obtaining a similar profile to the experimental absorption spectrum of the catalytic reaction mixture (see Fig. 2A and Fig. S16, ESI†). The most intense computed absorptions (higher oscillator strengths) appear centred in the interband transition region. We chose the three most intense excitations, which correspond to excited states 188, 232 and 247. For these electronic excitations we computed the electron–hole (e−–h+) (green-red) distributions to predict the electron density polarization in the corresponding excited states, representing the warm electrons and the hot deep holes expected for d–sp interband transitions (see Fig. 2E and Fig S17, ESI†). In all cases, a clear shift of the electron density from the inner part of the Au13 nanocluster towards the gold atom, positioned opposite to the location of the triple bond, is observed, together with a loss of electron density of the Au atoms close to the alkyne.
Fig. 2 depicts the MOs involved in the excited state 247, revealing a clear 5d gold composition of the occupied and the 6s/p composition of the virtual, in agreement with the proposed d–sp interband transitions, in which hot carriers were generated (see frontier MOs in Fig. S18, ESI†). In a further step, TDDFT optimizations of the three excited states 188, 232 and 247 were carried out to show the cluster distortion during the excitation process (see Fig. 2F). In these three cases, the observed changes reveal the elongation of Au–Au distances of the cluster on gold atoms localized in the opposite extreme of the cluster with respect to the triple bond (from 2.814 to 2.846 Å) and those stabilized by the phenyl group (from 2.911 to 2.947 Å). In contrast, the distances among gold atoms adjacent to the triple bond experienced a significant decrease (from 2.887 to 2.849 Å). These shifts in geometry suggest a slight detachment of three gold atoms from the Au13 cluster together with the alkyne molecule, consequently indicating the incipient formation of smaller gold SNCs from gold NPs in the excited state. This result shows that the visible light excitation of Au NPs in the presence of 5-phenyl-1-pentyne, gives rise to interband d–sp transitions for which the electron–hole generation favours the detachment of Au3 SNCs from the surface of the Au NPs, leading to a continuous feeding of these species in the reaction medium, further improving the catalytic efficiency.
In conclusion, this work demonstrates that white LED light irradiation can improve the efficiency of gold catalytic hydration of alkynes. We have developed a catalytic strategy in which a mixture of Au NPs and SNCs in a polymeric matrix can continuously feed the reaction with the catalytically active Au SNC species. We have also demonstrated that this photocatalytic process occurs as a result of the generation of hot carriers due to electronic d–sp interband transitions.
We acknowledge the DGI MICINN/FEDER (project number PID2022-139739NB-l00 (AEI/FEDER, UE)) and “ERDF A way of making Europe”. A. S. also acknowledges UR-CAR for a FPI grant.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Oliver-Meseguer and A. Leyva-Pérez, Recent Advances in Nanoparticle Catalysis, ed. P. W. N. M. Leeuwen and C. Claver, Springer, 2020, vol. 1, p. 1–37 Search PubMed.
- J. Cordón, G. Jiménez-Osés, J. M. López-De-Luzuriaga and M. Monge, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- R. Zhou, M. Shi, X. Chen, M. Wang and H. Chen, Chem. – Eur. J., 2009, 15, 4944–4951 CrossRef CAS PubMed.
- D. Bain, S. Maity, T. Debnath, A. K. Das and A. Patra, Mater. Res. Express, 2019, 6, 124004 CrossRef CAS.
- H. Duan and S. Nie, J. Am. Chem. Soc., 2007, 129, 2412–2413 CrossRef CAS PubMed.
- J. Oliver-Meseguer, A. Leyva-Pérez and A. Corma, ChemCatChem, 2013, 5, 3509–3515 CrossRef CAS.
- J. Oliver-Meseguer, A. Leyva-Pérez, S. I. Al-Resayes and A. Corma, Chem. Commun., 2013, 49, 7782–7784 RSC.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS PubMed.
- J. Oliver-Meseguer, I. Dominguez, R. Gavara, A. Doménech-Carbó, J. M. González-Calbet, A. Leyva-Pérez and A. Corma, Chem. Commun., 2017, 53, 1116–1119 RSC.
- Z. Zhang, C. Zhang, H. Zheng and H. Xu, Acc. Chem. Res., 2019, 52, 2506–2515 CrossRef CAS PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- Y. Zhang, T. Nelson, S. Tretiak, H. Guo and G. C. Schatz, ACS Nano, 2018, 12, 8415–8422 CrossRef CAS PubMed.
- P. Wang, A. V. Krasavin, M. E. Nasir, W. Dickson and A. V. Zayats, Nat. Nanotechnol., 2018, 13, 159–164 CrossRef CAS PubMed.
- J. U. Salmón-Gamboa, M. Romero-Gómez, D. J. Roth, A. V. Krasavin, P. Wang, W. Dickson and A. V. Zayats, Nanoscale Adv., 2021, 3, 767–780 RSC.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS PubMed.
- Q. Lin, S. Hu, T. Földes, J. Huang, D. Wright, J. Griffiths, B. de Nijs, E. Rosta and J. J. Baumberg, Sci. Adv., 2022, 8, eabp9285 CrossRef CAS PubMed.
- F. Mohammadparast, A. P. Dadgar, R. T. A. Tirumala, S. Mohammad, C. O. Topal, A. K. Kalkan and M. Andiappan, J. Phys. Chem. C, 2019, 123, 11539–11545 CrossRef CAS.
- W. Guo, A. C. Johnston-Peck, Y. Zhang, Y. Hu, J. Huang and W. D. Wei, J. Am. Chem. Soc., 2020, 142, 10921–10925 CrossRef CAS PubMed.
- B. Roche, T. Vo and W. S. Chang, Chem. Sci., 2023, 14, 8598–8606 RSC.
- J. Zhao, S. C. Nguyen, R. Ye, B. Ye, H. Weller, G. A. Somorjai, A. P. Alivisatos and F. Dean Toste, ACS Cent. Sci., 2017, 3, 482–488 CrossRef CAS PubMed.
- T. Fei Niu, D. Yun Jiang, S. Yuan Li, X. Ge Shu, H. Li, A. Ling Zhang, J. Yu Xu and B. Qing Ni, Tetrahedron Lett., 2017, 58, 1156–1159 CrossRef.
- X. Liu, L. Liu, Z. Wang and X. Fu, Chem. Commun., 2015, 51, 11896–11898 RSC.
- Y. Li, Z. Su, H. Hao and J. Zhuang, Chem. Commun., 2020, 56, 7669–7672 RSC.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Light: Sci. Appl., 2020, 9, 108 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02370f |
| This journal is © The Royal Society of Chemistry 2024 |