
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-throughput label-free opioid receptor binding assays using an automated desorption electrospray ionization mass spectrometry platform†
Yunfei
Feng‡
,
Nicolás M.
Morato‡
 ,
Kai-Hung
Huang
,
Mina
Lin
and
R. Graham
Cooks
*
,
Kai-Hung
Huang
,
Mina
Lin
and
R. Graham
Cooks
*
Department of Chemistry, Bindley Bioscience Center, and Purdue Institute for Cancer Research, Purdue University, West Lafayette, IN 47907, USA. E-mail: cooks@purdue.edu
First published on 10th July 2024
Abstract
The current opioid epidemic has incentivized the discovery of new non-addictive analgesics, a process that requires the screening of opioid receptor binding, traditionally performed using radiometric assays. Here we describe a label-free alternative based on high-throughput (1 Hz) ambient mass spectrometry for screening the receptor binding of new opioid analogues.
Opioids have been utilized as effective analgesics for over a hundred years. This is not surprising considering that the use of poppy extract (i.e. opium) can be traced back several millennia to the Sumerian and Egyptian civilizations.1 Recently, the over-prescription of opioids for pain treatment and the subsequent surge of their non-medical misuse, together with the increasing access to illicit and high-potency synthetic opioids (e.g. fentanyl and its analogues), have led to a public health crisis.2 Close to 650
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 opioid overdose deaths have been registered in the US alone since 1999, with more than half of those occurring since 2015.3 To address this crisis, public health authorities have initiated a multipronged strategy focusing on development of (i) new treatments for opioid addiction, (ii) overdose reversal interventions, and (iii) non-addictive therapeutic drugs for chronic pain.4
000 opioid overdose deaths have been registered in the US alone since 1999, with more than half of those occurring since 2015.3 To address this crisis, public health authorities have initiated a multipronged strategy focusing on development of (i) new treatments for opioid addiction, (ii) overdose reversal interventions, and (iii) non-addictive therapeutic drugs for chronic pain.4
In the context of drug discovery, extensive efforts have been directed towards new potent opioid antagonists as countermeasures against high-potency opioid overdoses and non-addictive opioid analgesics.4 An important initial parameter in assessing these classes of drug candidates is their binding affinity towards opioid receptors (ORs).5,6 As both OR agonists (e.g. morphine) and antagonists (e.g. naloxone) act through OR binding (the former further activate the receptor whereas the latter do not), the screening of binding affinities of new opioid drug candidates is thus fundamentally important prior to any further pharmacological characterization. Such screening traditionally takes place through a radiometric bioassay which uses the competitive displacement of radioligands (e.g. [3H] DAMGO, Tyr-D-Ala-Gly-N-MePhe-Gly-ol) from ORs in solution as a proxy for the binding strength of a drug candidate.7,8 Despite their effectiveness, radioactivity-based biological assays have inherent drawbacks to their implementation (e.g. safety concerns, cost, intensive labour requirements).
Here we demonstrate an alternative label-free and high-throughput approach for screening of OR binding by drug candidates using high-throughput chromatography-free mass spectrometry (MS), an approach that is increasingly utilized for drug discovery applications.9–11 Our particular system combines custom and commercial software, robotics, and analytical instrumentation, to allow automated assays and data handling.12 The core technology of the platform is desorption electrospray ionization (DESI), an ambient ionization method that provides rapid and direct MS analysis of complex samples in the open air.13–15 DESI uses a charged solvent spray to impact a sample on a surface, generating a thin film (typically ∼200 μm spot size) in which a microextraction event occurs facilitating desorption of material from the surface.13,14 The desorbed compounds are carried in secondary splashed microdroplets (generated through stochastic momentum transfer from the primary incoming spray) to the mass spectrometer.13,14 As they fly (ca. 100 ms) these secondary droplets undergo coulombic fission and desolvation to generate dry ions. Provided that the conditions are chosen appropriately, the ions are representative of the composition of the sampled surface.13,14 DESI is uniquely suited to carry out high-throughput biological assays as: (i) it is a contactless methodology that involves no sample transfer through capillaries, thus avoiding any potential clogging from standard non-volatile assay buffers which is a common problem in conventional MS approaches, (ii) the key online extraction event facilitates the analysis of complex samples, (iii) it provides spatial control over the sampling event, so spatially distributed samples on a surface can be rapidly analysed by moving them sequentially under the spray.14,16
Leveraging these advantages, high-throughput DESI-MS analysis is achieved using high-precision robotics including a fluid handling workstation and an articulated robot arm to generate high-density arrays of samples spotted onto PTFE-coated glass slides (up to 6144 spots per slide, each spot being ca. 800 μm in diameter and separated by 1.125 mm centre-to-centre at the highest density). Once spotted, the slides are robotically transferred to the DESI stage of a mass spectrometer for automated analysis by oscillating over each spot and then jumping from spot-to-spot, with an effective analysis time of 500 ms and an overall analysis throughput of ca. 1 Hz. The size difference between the DESI spray and the sample spot, together with the porosity of the PTFE membrane and the optimized geometry of the DESI source, guarantees the absence of cross-contamination between samples. The spray cross section is much smaller than the analyte spot size so that the small amounts of material deposited (here 150 nL) are always under-sampled. The complete high-throughput experiment, including data analysis, is completed automatically using a combination of Python- and MATLAB-based custom software.12,17
To assess OR binding, the fluid handling workstation is used to set up a label-free competitive binding assay with commercial membrane-bound preparations of either μ or δ human ORs. In both cases, leucine enkephalin (LeuEnk; Tyr-Gly-Gly-Phe-Leu) is used as competitive ligand, as it is an endogenous opioid peptide with agonistic activity on both μ or δ ORs with dissociation constants (KD) around 8 and 5 nM, respectively.18,19 In this assay, as in traditional radiometric methods, the extent of binding of the competitive ligand is used as a proxy for the binding affinity of a test compound of interest. The DESI-MS assay measures the concentration of unbound LeuEnk after equilibrium is reached in the presence of the test compound, a value that can be more robustly determined compared to the significantly lower amount of LeuEnk that is bound to the OR. Separation of the bound and unbound ligand can be achieved readily using 1-μm filters, an operation that can be parallelized and automated using 96-well format filtration plates. After filtering and washing, a volumetric aliquot of the solution is spiked with DADLE (Tyr-D-Ala-Gly-Phe-D-Leu), a synthetic LeuEnk analogue used as internal standard for DESI-MS quantitation (Fig. 1A and Fig. S1, ESI†). The ratio of LeuEnk ([M–H]−m/z 554) to DADLE ([M–H]−m/z 568), determined via high-throughput DESI-MS (Fig. 1D), reflects the concentration of free LeuEnk in the assay solution. Note that this competitive binding assessment approach is advantageous as it allows quantitative DESI-MS to exclusively target LeuEnk and DADLE regardless of the test compound structure or concentration. Thus, targeted optimization of the instrumental conditions (in our case using quadrupole mass filtering and target enhancement via synchronization of the time-of-flight conditions), together with the intrinsic advantages of DESI for complex sample analysis, provides very high sensitivity (ca. 200 fg limit of quantitation) despite the fast analysis (1 sample per second) performed directly from the complex bioassay matrix (non-volatile buffer).
 | ||
| Fig. 1 Workflow and validation of the label-free DESI-MS competitive OR binding assays. Assays are automatically prepared in plate format and, after incubation, filtration, and internal standard (i.e. DADLE) addition, they are spotted in high-density arrays that are directly analysed via DESI-MS (A). This method was initially validated by assessing LeuEnk binding as a function of OR concentration (B) and (C). Free LeuEnk quantitation proceeded using its ion intensity relative to DADLE from averaged data (ca. 500 ms, equivalent to 5 scans) full scan mass spectra (D). | ||
The high-throughput label-free DESI-MS methodology was validated by assessing LeuEnk binding upon incubation over a range of μ and δ OR concentrations (5–200 μg mL−1 of commercial membrane-bound OR preparations). As shown in Fig. 1B and C, the free LeuEnk concentration decreases significantly as the concentration of OR increases, and the dose–response behaviour obtained reflects the slightly higher binding affinity of LeuEnk towards δ OR compared to μ OR. For compound screening, assay results are expressed as a normalized response which considers the measured ratios for controls run in parallel with each batch of experiments. Normalization was deemed necessary as batch-to-batch variation in the OR preparations was found to be significant. This normalized assay response follows the fraction of free LeuEnk relative to bound LeuEnk in the absence of a test compound (minimum assay response, topFig. 1D), corrected by the amount bound via non-specific phenomena (maximum assay response, bottomFig. 1D). A large assay response indicates a high relative concentration of free LeuEnk and thus a strong binding of the test compound to the OR, as LeuEnk is significantly displaced by the test compound.
The performance of the method was evaluated by assessing the dose–response binding of multiple drug compounds towards both μ and δ ORs (Fig. 2). For each receptor a suite of know agonists, antagonists, and nonopioids analgesics were tested.
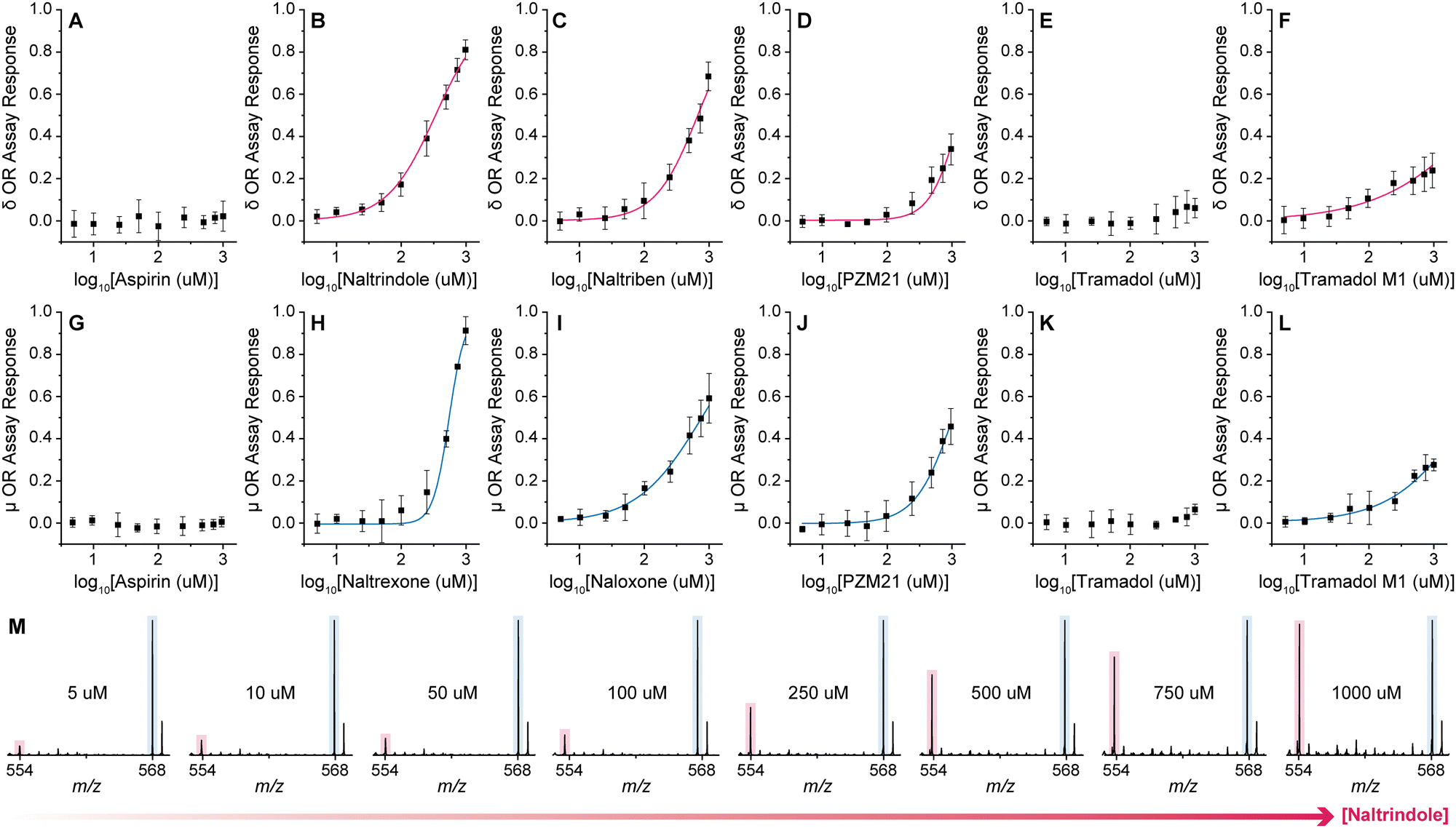 | ||
| Fig. 2 Dose–response OR binding results for a suite of compounds including OR agonists, antagonists, and non-opioids. The top curves (A)–(F) show compounds assessed against δ OR whereas the curves below (G)–(L) show the results for μ OR. All data points represent the average of at least 3 independent experiments with at least 24 technical replicates. Error bars indicate standard deviations. Representative spectra corresponding to the dose–response experiments for naltrindole and δ OR are also included (M). | ||
Overall, the assay response trends obtained with the label-free DESI-MS approach agree well with literature data based on radiometrically estimated Kd values (Table S1, ESI†). For instance, between the two μ OR antagonists, naltrexone was found to be more potent than naloxone,19,20 as was naltrindole compared to naltriben in the case of δ OR.21,22 PZM21, an experimental OR agonist, showed binding to both receptors, with higher affinity for the μ OR, and in all cases lower activity than any of the four antagonists.23 Tramadol showed almost negligible binding towards both ORs with significantly lower affinity than its main metabolite (M1) O-desmethyltramadol, which is well known as its active form.24,25 Aspirin, as expected, showed no binding as it reduces pain by acting on cyclooxygenases rather than ORs.26
Based on the agreement of this label-free bioassay approach with reported bioactivity trends, we proceeded to demonstrate its integration with the already established capability of automated high-throughput DESI-MS for organic reaction screening.27,28 This combination is relevant in the development of new opioids as, for instance, the screening of late-stage functionalization (LSF) reactions to generate new analogues of bioactive compounds is a common iterative step for hit optimization towards drug leads.27,29 To showcase this integrated MS-based approach, we utilized the results of a screening campaign for the LSF of PZM21 (1) targeted towards selective modifications of its phenolic moiety through click-like reactions via sulphur(VI) fluoride exchange (SuFEx) and ene-type chemistry.27 This screening was carried out without any reaction incubation, so leveraging another unique advantage of the DESI methodology related to the acceleration of reactions at the surface of the secondary microdroplets containing desorbed reactants.27,30 Such acceleration has been related to the partial solvation of reagents as well as the high electric field at the droplet-air interface which generates reactive species and extremes of pH.31–33 A selection of successful reactions observed through microdroplet-based screening was chosen for scale up in batch for bioactivity testing (Fig. 3). Overall, no significant change in OR binding was observed upon fluorosulfurylation (2), complete SuFEx (3) or ene-like derivatization (4) of the PZM21 phenol, an observation that can be ascribed to the flexible environment of the receptor around that moiety and the likely preservation of important hydrogen bonding interactions.23,34
 | ||
| Fig. 3 Structures and activity of PZM21 analogues selected from LSF screening campaign. Three functionalized compounds (2–4), see (A) were selected for OR binding assessment and compared against PZM21 (1). No significant differences in binding were observed in the screening (B), an observation that was confirmed via dose–response studies for 4 (C). | ||
The demonstration of label-free binding assays to receptors adds to the bioanalytical toolbox of automated high-throughput DESI-MS in the drug discovery space. Previously established capabilities include the analysis of biosamples for biomarker discovery or large-cohort validation,35 and the characterization and screening of enzymatic targets.16,36 We anticipate that these capabilities, together with those for reaction screening (as shown here) and nanoscale synthesis,37 together with predictive computational tools,38,39 will allow consolidation of the early drug discovery workflow around a single technology.
This work was supported by NCATS through the ASPIRE Reduction-to-Practice Challenge and the UG3 grant TR004139, as well as by Waters Corporation (grant 40002775). The authors acknowledge the use of the Purdue Make It Platform in the Metabolite Profiling Facility of the Bindley Biosciences Center, a core facility of the NIH-funded Indiana CTSI. The authors also acknowledge support from the Purdue Institute for Cancer Research, NIH grant P30 CA023168, and greatly appreciate the technical support of Dr Steven Pringle (Waters Corporation) as well as assistance of Zihan Qu in the purification and characterization of the PZM21 analogues.
Data availability
All data is available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Katz, Clin. J. Pain, 2007, 23, 303–306 CrossRef PubMed.
- Y. K. Lee, M. S. Gold, K. Blum, P. K. Thanos, C. Hanna and B. S. Fuehrlein, Front. Public Health, 2024, 11, 1274719 CrossRef PubMed.
- E. J. Stringfellow, T. Y. Lim, K. Humphreys, C. DiGennaro, C. Stafford, E. Beaulieu, J. Homer, W. Wakeland, B. Bearnot, R. K. McHugh, J. Kelly, L. Glos, S. L. Eggers, R. Kazemi and M. S. Jalali, Sci. Adv., 2022, 8, eabm8147 CrossRef CAS PubMed.
- N. D. Volkow and F. S. Collins, N. Engl. J. Med., 2017, 377, 391–394 CrossRef PubMed.
- T. Che and B. L. Roth, Annu. Rev. Biochem., 2021, 90, 739–761 CrossRef CAS PubMed.
- A. Manglik, Biol. Psychiatry, 2020, 87, 6–14 CrossRef CAS PubMed.
- R. N. DeHaven, J. A. Cassel, R. T. Windh and D. L. DeHaven-Hudkins, Curr. Protoc. Pharmacol., 2005, 29, 1.4.1 Search PubMed.
- C. A. Flanagan, GPCR-radioligand binding assays, Elsevier, 2016, vol. 132 Search PubMed.
- Y. Xin, S. W. Foster, D. M. Makey, D. Parker, J. Bradow, X. Wang, S. Berritt, R. Mongillo, J. P. Grinias and R. T. Kennedy, Anal. Chem., 2024, 96, 4693–4701 CrossRef CAS PubMed.
- F. Pu, N. L. Elsen and J. D. Williams, ACS Med. Chem. Lett., 2020, 11, 2108–2113 CrossRef CAS PubMed.
- H. Hu, A. N. Singh, D. Lehnherr, V. Mdluli, S. W. Chun, A. M. Makarewicz, J. R. Gouker, O. Ukaegbu, S. Li, X. Wen, D. G. McLaren, J. E. Velasquez, J. C. Moore, S. Galanie, E. Appiah-Amponsah and E. L. Regalado, Anal. Chem., 2024, 96, 1138–1146 CrossRef CAS PubMed.
- N. M. Morato, M. T. Le, D. T. Holden and R. Graham Cooks, SLAS Technol., 2021, 26, 555–571 CrossRef CAS PubMed.
- Z. Takáts, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef PubMed.
- N. M. Morato and R. G. Cooks, Acc. Chem. Res., 2023, 56, 2526–2536 CrossRef CAS PubMed.
- C. Yan, F. Parmeggiani, E. A. Jones, E. Claude, S. A. Hussain, N. J. Turner, S. L. Flitsch and P. E. Barran, J. Am. Chem. Soc., 2017, 139, 1408–1411 CrossRef CAS PubMed.
- N. M. Morato, D. T. Holden and R. G. Cooks, Angew. Chem., Int. Ed., 2020, 59, 20459–20464 CrossRef CAS PubMed.
- T. J. P. Sobreira, L. Avramova, B. Szilagyi, D. L. Logsdon, B. P. Loren, Z. Jaman, R. T. Hilger, R. S. Hosler, C. R. Ferreira, A. Koswara, D. H. Thompson, R. G. Cooks and Z. K. Nagy, Anal. Methods, 2020, 12, 3654–3669 RSC.
- F. Wieberneit, A. Korste, H. B. Albada, N. Metzler-Nolte and R. Stoll, Dalton Trans., 2013, 42, 9799 RSC.
- L. Toll, I. P. Berzetei-Gurske, W. E. Polgar, S. R. Brandt, I. D. Adapa, L. Rodriguez, R. W. Schwartz, D. Haggart, A. O’Brien, A. White, J. M. Kennedy, K. Craymer, L. Farrington and J. S. Auh, NIDA Res. Monogr., 1998, 178, 440–466 CAS.
- R. J. Valentino, J. L. Katz, F. Medzihradsky and J. H. Woods, Life Sci., 1983, 32, 2887–2896 CrossRef CAS PubMed.
- P. S. Portoghese, H. Nagase, K. E. MaloneyHuss, C. E. Lin and A. E. Takemori, J. Med. Chem., 1991, 34, 1715–1720 CrossRef CAS PubMed.
- P. C. Contreras, L. Tam, E. Drower and M. F. Rafferty, Brain Res., 1993, 604, 160–164 CrossRef CAS PubMed.
- A. Manglik, H. Lin, D. K. Aryal, J. D. McCorvy, D. Dengler, G. Corder, A. Levit, R. C. Kling, V. Bernat, H. Hübner, X.-P. Huang, M. F. Sassano, P. M. Giguère, S. Löber Da Duan, G. Scherrer, B. K. Kobilka, P. Gmeiner, B. L. Roth and B. K. Shoichet, Nature, 2016, 537, 185–190 CrossRef CAS PubMed.
- J. Lai, S. Ma, F. Porreca and R. B. Raffa, Eur. J. Pharmacol., 1996, 316, 369–372 CrossRef CAS PubMed.
- H. Potschka, E. Friderichs and W. Löscher, Br. J. Pharmacol., 2000, 131, 203–212 CrossRef CAS PubMed.
- B. Kaur and P. Singh, Bioorg. Chem., 2022, 121, 105663 CrossRef CAS PubMed.
- K. Huang, N. M. Morato, Y. Feng and R. G. Cooks, Angew. Chem., Int. Ed., 2023, 62, e202300956 CrossRef CAS PubMed.
- M. Wleklinski, B. P. Loren, C. R. Ferreira, Z. Jaman, L. Avramova, T. J. P. Sobreira, D. H. Thompson and R. G. Cooks, Chem. Sci., 2018, 9, 1647–1653 RSC.
- R. Poulain, D. Horvath, B. Bonnet, C. Eckhoff, B. Chapelain, M.-C. Bodinier and B. Déprez, J. Med. Chem., 2001, 44, 3378–3390 CrossRef CAS PubMed.
- M. Girod, E. Moyano, D. I. Campbell and R. G. Cooks, Chem. Sci., 2011, 2, 501–510 RSC.
- L. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2024, 63, e202400118 CrossRef CAS PubMed.
- H. Xiong, J. K. Lee, R. N. Zare and W. Min, J. Phys. Chem. Lett., 2020, 11, 7423–7428 CrossRef CAS PubMed.
- M. A. Mehrgardi, M. Mofidfar and R. N. Zare, J. Am. Chem. Soc., 2022, 144, 7606–7609 CrossRef CAS PubMed.
- Z. Zhao, T. Huang and J. Li, Int. J. Mol. Sci., 2020, 21, 4699 CrossRef CAS PubMed.
- N. M. Morato, H. M. Brown, D. Garcia, E. H. Middlebrooks, M. Jentoft, K. Chaichana, A. Quiñones-Hinojosa and R. G. Cooks, Sci. Rep., 2022, 12, 18851 CrossRef CAS PubMed.
- S. C. Kulathunga, N. M. Morato, Q. Zhou, R. G. Cooks and A. D. Mesecar, ChemMedChem, 2022, 17, e202200043 CrossRef CAS PubMed.
- R. G. Cooks, Y. Feng, K. Huang, N. M. Morato and L. Qiu, Isr. J. Chem., 2023, 63, e202300034 CrossRef CAS PubMed.
- W. Mangione, Z. Falls and R. Samudrala, Front. Pharmacol., 2023, 14, 1113007 CrossRef CAS PubMed.
- Z. Tu, T. Stuyver and C. W. Coley, Chem. Sci., 2023, 14, 226–244 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, supporting literature Kd values and calculations. See DOI: https://doi.org/10.1039/d4cc02346c |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |