
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst development for O2-assisted oxidative dehydrogenation of propane to propylene
Huimin
Liu
 *a,
Shaoyuan
Sun
a,
Dezheng
Li
a and
Yiming
Lei
*b
*a,
Shaoyuan
Sun
a,
Dezheng
Li
a and
Yiming
Lei
*b
aSchool of Chemical and Environmental Engineering, Liaoning University of Technology, Jinzhou, 121001, Liaoning Province, P. R. China. E-mail: liuhuimin08@tsinghua.org.cn
bDepartament de Química (Unitat de Química Inorgànica), Facultat de Ciències, Universitat Autònoma de Barcelona (UAB), Cerdanyola del Valles, 08193, Barcelona, Spain. E-mail: yiming.lei@uab.cat
First published on 24th June 2024
Abstract
O2-Assisted oxidative dehydrogenation of propane (O2-ODHP) could convert abundant shale gas into propylene as an important chemical raw material, meaning O2-ODHP has practical significance. Thermodynamically, high temperature is beneficial for O2-ODHP; however, high reaction temperature always causes the overoxidation of propylene, leading to a decline in its selectivity. In this regard, it is crucial to achieve low temperatures while maintaining high efficiency and selectivity during O2-ODHP. The use of catalytic technology provides more opportunities for achieving high-efficiency O2-ODHP under mild conditions. Up to now, many kinds of catalytic systems have been elaborately designed, including transition metal oxide catalysts (such as vanadium-based catalysts, molybdenum-based catalysts, etc.), transition metal-based catalysts (such as Pt nanoclusters), rare earth metal oxide catalysts (especially CeO2 related catalysts), and non-metallic catalysts (BN, other B-containing catalysts, and C-based catalysts). In this review, we have summarized the development progress of mainstream catalysts in O2-ODHP, aiming at providing a clear picture to the catalysis community and advancing this research field further.
 Huimin Liu | Huimin Liu is a professor at Liaoning University of Technology in the School of Chemical and Environmental Engineering. She received her PhD degree from Tsinghua University, China (2013), and then joined Kansai University (2013–2014), National Institute for Materials Science (2014–2017), and University of Sydney (2017) as a post-doctoral researcher. Her research interests are photochemistry, environmental chemistry, and heterogeneous catalyst design. |
 Yiming Lei | Yiming Lei received his MS degree from Tianjin University in 2022. In Jan. 2023, he joined Professor Liu's group as a Research Assistant at Liaoning University of Technology, China. Now, he is a PhD student in the Department of Chemistry at Universitat Autònoma de Barcelona, Spain. His current research focuses on the functionalization of the 2D germanene material for various practical applications such as (bio)sensor and photo/electrocatalysis, as well as the methane conversion and carbon dioxide reduction technologies. |
1. Introduction
As one of the most important basic petrochemical raw materials, propylene can be used to produce various important organic chemicals, such as acrylonitrile, epichlorohydrin, epichlorohydrin, isopropanol, acetone, acrylic esters, etc.1–4 With the outbreak of the shale gas revolution, the use of propane extracted from shale gas as raw material to prepare propylene can not only optimize the utilization of its low-carbon alkanes and by-products but also produce important basic organic chemical raw material, which has important practical significance.5–7 Considering the fact that there are abundant shale gas resources in the world, it is expected that there will be sufficient shale gas production to cope with the significant supply–demand gap for propylene.8The emergence of the catalytic energy conversion technique is one of the reliable strategies,9–13 which can achieve the conversion of shale gas into propylene. Propane can undergo thermal cracking to release hydrogen and produce propylene under anaerobic conditions (direct dehydrogenation of propane), eqn (1).14,15 With the assistance of suitable catalysts, at a reaction temperature of 590–630 °C, the one-way conversion rate of propane can reach 33–44%, and the selectivity of propylene is above 80%.14,15 Although catalytic propane conversion is very promising and attractive from a thermodynamic view, the deep dehydrogenation of propane tends to form carbon deposition, which in turn leads to catalyst deactivation.16,17
Owing to the above problem, the O2-assisted oxidative dehydrogenation process using propane as raw material and O2 as a strong oxidant (oxidative dehydrogenation of propane (O2-ODHP), eqn (2)) has attracted significant attention and has been considered as another effective process for producing propylene due to its remarkable cost advantage.18–21 ODHP is a reversible and strongly endothermic reaction. Theoretically, the equilibrium constant of O2-ODHP should increase with the increase of reaction temperature, but practically, its equilibrium constant still remains low at high temperatures. Therefore, from a thermodynamic perspective, O2-ODHP needs to be carried out at high temperatures and low pressure. However, the high reaction temperature conditions intensify the overoxidation of propylene, leading to a decrease in the selectivity of desired propylene. In this case, the fabrication of well-designed catalysts with tunable selectivity and high activity is important to directionally obtain the target product propylene from O2-ODHP at low temperatures.
| C3H8 → C3H6 + H2 (+Coke) | (1) |
| C3H8 + 1/2O2 → C3H6 + H2O | (2) |
To date, a great number of catalysts have been developed for O2-ODHP (Scheme 1), including transition metal oxide catalysts (such as vanadium-based catalysts, molybdenum-based catalysts, etc.), transition metal-based catalysts (such as Pt nanoclusters), rare earth metal oxide catalysts (especially CeO2 related catalysts) and non-metallic catalysts (BN, other B-containing catalysts and C-based catalysts). Herein, the recent progress of these catalysts in O2-ODHP is summarized and the underlying mechanism is revealed, with the aim to pave pathways for the rational design of more efficient catalysts for ODHP and advance this research field further.
 | ||
| Scheme 1 Mainstream catalysts for O2-ODHP. | ||
2. Reaction mechanism in ODHP reaction
2.1. O2-Assited ODHP reaction
O2-ODHP attracts wide attention since it can induce chemical equilibrium by consuming H2. Although the reaction routes might be different over diverse catalysts, some common features in the O2-ODHP reaction mechanism could be summarized as follows: (i) interactions of C3H8 with the catalyst surface (weak/physical adsorption); (ii) dissociation of the C–H bond, leading to the formation of intermediate species; (iii) interaction of intermediates with adjacent surface oxygen and formation of C3H6; and (iv) cyclic reduction/reoxidation of the catalyst.22 During the O2-ODHP reaction, the coordination number of active centers will affect the ODHP process. For instance, high coordination number is beneficial for the insertion of oxygen species into C3H8 to generate partially oxygenated products or carbon oxides.23 Meanwhile, the local environment of active centers can change between several configurations, suggesting that the catalytic O2-ODHP might be related to the crystal structures of different types of catalysts.24 The strength and reducibility of the oxygen ions also play an important role. What's more, the acid and/or basic centers on catalysts also affect the formation rate of olefinic intermediates and the desorption rate of products.In the conventional ODHP reaction, the conversion of propane into C3H8 is endothermic and volume-increasing, meaning the conditions with high temperatures and low pressures are beneficial for achieving a high propane conversion rate. In practical industrial systems, the ODHP process is always conducted at 550–700 °C to achieve appreciable propylene yields.25 However, the high temperature leads to side or competitive reactions, forming undesired C2H4 and CH4 products due to C–C cracking. Meanwhile, coke is also one of the products from these side reactions. Therefore, it is necessary to consider the ability to dissociate C–H and avoid C–C bond break during the catalyst design steps.26
In order to selectively oxidize C3H8 to C3H6 and H2/H2O (eqn (1) and (2)), O2 as an oxidant has several advantages including low reaction temperatures, resisting coke formation, and extending catalyst longevity.27,28 The main challenge, as mentioned above, is the overoxidation reactions, which oxidize C3H8 to CO and useless CO2. In this review, the representative and recent works on catalyst design for ODHP using O2 as the oxidant will be focused on, discussing the feasible strategies that can improve selectivity and stability under mild conditions.
Although some excellent reviews have summarized the catalytic systems for ODHP, they have focused on the relationship between ODHP performance and catalyst structures.29–31 Other reviews have paid much attention to ODHP with CO2 assistance32 or the comparison of direct and CO2-assisted ODHP (CO2-ODHP).26 Thus, the regulation of O2-ODHP selectivity under mild conditions has not received enough attention. Considering the fact that ODHP technology could convert abundant shale gas into propylene, which is an important chemical raw material, and thus has practical significance, the representative works about utilizing catalyst design to improve the O2-ODHP selectivity should be systematically reviewed to encourage more in-depth research in this area in the future.
2.2. CO2-Assisted ODHP reaction
CO2-ODHP (C3H8 + CO2 → C3H6 + CO + H2O) is able to inhibit the formation of CO2 by consuming H2via the reverse water–gas shift reaction (RWGS, CO2 + H2 → CO + H2O).33 Besides the desired C3H6 and value-added syngas (CO and H2) products, the coke from the dehydrogenation process can be removed via the reverse Boudouard reaction (CO2 + C → 2CO). Hence, it is possible to perform CO2-assisted ODHP under high-temperature conditions with high efficiency and stability.The mainstream mechanisms for CO2-assisted ODHP are a direct route with a redox mechanism and an indirect route combining ODHP and RWGS reactions.26 For the former, C3H8 interacts with O species to form C3H6 and water, and generates O vacancies over the catalyst surface. Then, CO2 refills the O vacancies, supplying active O species and producing CO simultaneously. The direct route can be completed at a single active site or a dual site. Regarding the indirect route, ODHP and RWGS reactions could be completed to realize the simultaneous conversion of CO2 and C3H8. During this pathway, the coke should be rapidly removed via the reverse Boudouard reaction.
Many research results about CO2-ODHP have been published. For instance, Furukawa et al. reported a Pt–Co–In ternary nanoalloy on CeO2 with (Pt1−xCox)2In3 pseudo-binary alloy structure.34 The synergistic effect of alloying platinum with indium and cobalt leads to high activity involving C3H6 selectivity and CO2 reduction ability. The existence of Co species offers a high density of states near the Fermi level, reducing the energy barrier of CO2 reduction. The cooperation between the alloy with CO2 activation ability and CeO2 with oxygen releasing ability facilitates coke combustion, thereby enhancing the catalyst stability. Song's group reported an efficient bifunctional CO2-ODHP site capable of activating C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds with inhibited C–C scission capacity.35 In detail, their work proposed a La-modified binuclear Feoxo (La-Feoxo) site stabilized on a silicalite-1 support, achieving 32.7% C3H8 conversion with 83.2% C3H6 selectivity. The improvement should be attributed to the balance in the acid–base property due to FeOx dispersion and La modification. The elaborate active site could stabilize HCOO* intermediates toward enhanced CO2-assisted H removal efficiency.
O bonds with inhibited C–C scission capacity.35 In detail, their work proposed a La-modified binuclear Feoxo (La-Feoxo) site stabilized on a silicalite-1 support, achieving 32.7% C3H8 conversion with 83.2% C3H6 selectivity. The improvement should be attributed to the balance in the acid–base property due to FeOx dispersion and La modification. The elaborate active site could stabilize HCOO* intermediates toward enhanced CO2-assisted H removal efficiency.
At present, the main problem during CO2-ODHP is the dry reforming of propane (DRP) competing reaction (C3H8 + 3CO2 → 6CO + 4H2). The DRP reaction could be the dominant reaction under high-temperature conditions, converting too much C3H8 into CO instead of C3H6.36 Therefore, to avoid the undesired DRP process, the C–H/C–C scission ability of C3H8 should be mainly focused on during the development of CO2-ODHP catalysts.37 A few works have summarized and discussed the soft oxidant-assisted ODHP system from three aspects: reaction mechanisms, catalyst composition, and oxidant footprint analysis. Interested readers are recommended to read ref. 29 and 34.
2.3. Chemical looping ODHP reaction
Although propane conversions are promising to get high-value-added chemicals, the traditional ODHP still has inevitable disadvantages, especially in the aspects of safety, high operation, and investment costs.38 For example, when feeding C3H8 and O2, the mixtures might evenly cause a potential explosion.39 What's more, it is difficult to separate specific products from the product stream in traditional ODHP reactors, leading to high requirements for operation and equipment.40 Although CO2 as a soft oxidant has been developed to avoid oxidation, this weak oxidant also has its own limitations, such as high price as feedstock and downstream separation.41,42In order to overcome the above disadvantages, chemical looping ODHP (CL-ODHP) combining dehydrogenation of propane and catalyst regeneration is explored and used in industries.43 In a CL-ODHP system, the reaction routes include (i) the conversion of propane into propylene and water via active oxygen species (oxygen carrier) from lattice oxygen (C3H8 + MOx → C3H6 + MOx−1 + H2O); (ii) transfer of the reduced oxygen carrier to the air reactor, where the missing lattice oxygen of the active phase is re-supplied by the air (MOx−1 + 1/2O2 → MOx); and (iii) transfer of the re-oxidized oxygen carriers to ODHP reactors to complete the chemical loop. Therefore, through repeated reduction and oxidation of active species, the oxygen can migrate from the air reactor to the ODHP reactor and take part in the CL-ODHP process. The advantages of CL-ODHP include (i) the decline in energy consumption by H2 oxidation; (ii) reduced investment and operation cost due to the elimination of separation equipment; (iii) avoidance of the potential explosion of C3H8; and (iv) reduced CO2 emissions by indirect flameless combustion of H2.44,45
The development of a suitable catalyst for CL-ODHP is crucial, but compared to the other two ODHP reaction systems, the investigations of catalyst design in the CL-ODHP field are much less. For instance, Gong's group designed multifunctional ferric vanadate–vanadium oxide (FeVO4–VOx) redox catalysts by combining chemical looping-selective H2 combustion and ODHP. VOx provided dehydrogenation sites, and FeVO4 acted as an oxygen carrier for H2 combustion. After 200 chemical looping cycles for the re-oxidation of FeVO4, FeVO4–VOx could maintain an integral performance of 81.3% propylene selectivity at 42.7% propane conversion. More importantly, the authors proposed a hydrogen spillover-mediated coupling mechanism. They believed that the H species generated at VOx sites could transfer to adjacent FeVO4 for combustion, leading to the formation of C3H6. The author's group also reported a MoO3–Fe2O3 catalyst with a similar CL-ODHP mechanism.46 Through introducing Mo species onto Fe2O3, the MoO3–Fe2O3 catalyst achieved very stable catalytic behaviour after 300 cycles with ∼49% propane conversion and ∼90% propylene selectivity. Several works have comprehensively discussed the recent development of the CL-ODHP system,39,47,48 and therefore the subsequent sections will mainly focus on O2-assisted ODHP, which has not been systematically summarized in recent years.
3. Transition metal oxide catalysts
Transition metal oxide (TMO) catalysts, including vanadium-, molybdenum-, cobalt-, and nickel-based catalysts, mixed transition metal oxide catalysts, etc., are excellent catalysts for O2-ODHP because incompletely filled d-orbitals allow TMO to both donate and accept electrons easily from other molecules.49 The real implementation of these catalysts in O2-ODHP is reviewed in this sub-section.3.1. Vanadium oxide-based catalysts
In the past several decades, vanadium-based catalysts have been the most extensively studied among transition metal oxide catalysts.27,50 It has been reported that there are several strategies to improve the catalytic performance of vanadium-based catalysts in O2-ODHP, mainly including (1) optimizing the distribution and amount of active site VOx, (2) selecting a suitable catalyst support, and (3) introducing highly efficient promoters.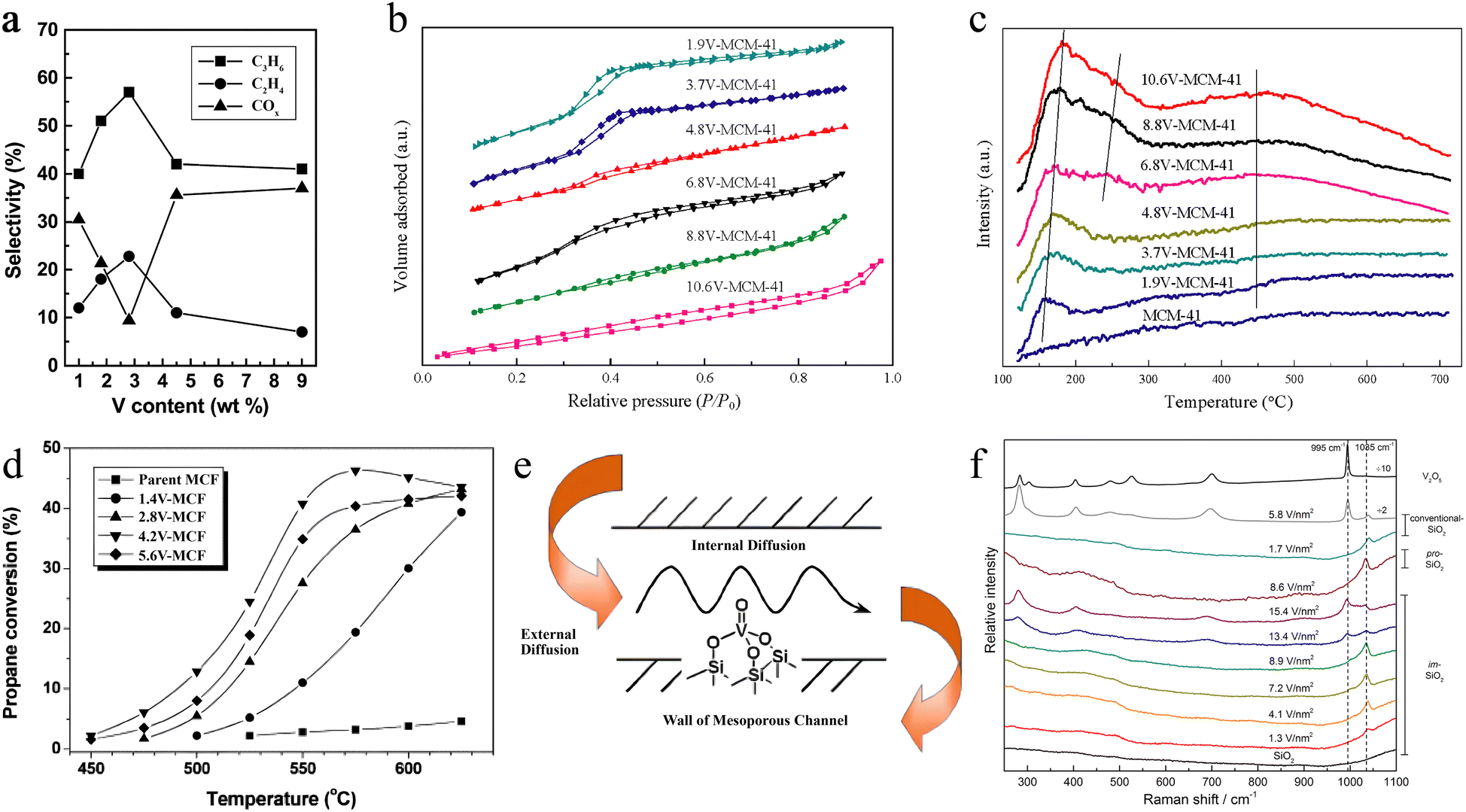 | ||
Fig. 1 (a) Variation of selectivity to propylene (■), ethylene (●), and COx (▲) with vanadium surface densities. Reaction temperature = 600 °C; propane conversion = 40%.52 Copyright 2004 Elsevier. (b) N2 adsorption–desorption isotherms of nV-MCM-41 samples. (c) NH3-TPD profiles of nV-MCM-41 samples.55 Copyright 2018 Elsevier B.V. (d) Propane conversion as a function of temperature on V-MCF catalysts. (Reaction conditions: C3H8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:N2 = 1:1:8; GHSV = 72000 L kgcat−1 h−1.) (e) Schematic illustration of the molecular transport effect on V-containing mesoporous catalysts during the oxidative dehydrogenation of propane.54 Copyright 2005 Elsevier. (f) Raman spectra of dehydrated vanadia catalysts supported on im-SiO2, pro-SiO2, and conventional-SiO2, compared to the Raman spectra of im-SiO2 (bottom) and bulk V2O5 (top).58 Copyright 2015 American Chemical Society. :O2:N2 = 1:1:8; GHSV = 72000 L kgcat−1 h−1.) (e) Schematic illustration of the molecular transport effect on V-containing mesoporous catalysts during the oxidative dehydrogenation of propane.54 Copyright 2005 Elsevier. (f) Raman spectra of dehydrated vanadia catalysts supported on im-SiO2, pro-SiO2, and conventional-SiO2, compared to the Raman spectra of im-SiO2 (bottom) and bulk V2O5 (top).58 Copyright 2015 American Chemical Society. | ||
For regulating the dispersion of V active sites, Guo's group designed mesoporous nV-MCM-41 molecular sieves via surface engineering.55 Compared to pristine V2O5, at low concentrations in nV-MCM-41, the V species existed in an amorphous state and became incorporated into the silicate framework or dispersed on mesoporous channels. The N2 adsorption–desorption isotherms showed typical type-IV isotherms, suggesting the maintenance of the mesoporous structure after the addition of V species (Fig. 1b). The NH3-TPD characterization was carried out to investigate the acidic characteristics. The results suggested that the isolated tetrahedral-like coordinated VOx species had the properties of Lewis acid sites, which could be adjusted via changing the concentration of V elements (Fig. 1c). The optimal propane conversion rate could reach 58% accompanied by a propene selectivity of ∼92% on 6.8 V-MCM-41. The authors believed that the improvement on catalytic efficiency and selectivity should be ascribed to the large specific surface of MCM-41 with a highly ordered mesoporous structure. But the increasing number of acidic sites reduced propene release from the V active sites, leading to deep oxidation and carbon deposition. This work also confirmed that suitably controlling the number and dispersion of VOx sites on the substrate is a crucial factor for achieving efficient and selective O2-ODHP reaction.
Another example of regulating activity and selectivity via utilizing the morphology and specific absorption sites on supports was reported by Fan et al.54 Fan's group loaded vanadium on MCF at a loading amount of 4.2% and evaluated the catalytic performance in O2-ODHP. Under the reaction temperature of 550 °C and atmospheric pressure conditions, the conversion of propane reached 40.8%, the selectivity of propylene was 68.5%, and the one-way yield was as high as 27.9% (Fig. 1d). The authors suggested that the larger specific surface area and the uniform pore size of the support MCF were beneficial for the rational dispersion of catalytic active sites (VOx, Fig. 1e). Meanwhile, the weak acidity of MCF also promoted the selectivity of propylene.
Considering that the catalytic performance of supported VOx is often influenced by its two- or three-dimensional dispersion, Grant et al. used an SiO2 inert support (im-SiO2) with low reactivity of surface SiOH groups toward the precursor to avoid undesired three-dimensional nanoparticles, which were often formed on conventional supports like Al2O3, TiO2, Nb2O5, or ZrO2.58 Raman spectra suggested that im-SiO2 did not show any sharp signal at 995 cm−1 from 3D V2O5, meaning the enhanced 2D dispersion behavior of V2O5 species (Fig. 1f). Because of the strong structure-sensitivity of O2-ODHP, 2D V2O5 loaded im-SiO2 had faster C3H8 consumption rate and higher TOF. Their results offered new opportunities for SiO2-supported V-based oxides and even other metal oxides applied for O2-ODHP.
Regarding the above vanadium-based catalysts, it is well accepted that their intrinsic activities originate from the strong redox properties of V5+ species. In O2-ODHP, they follow the Mars–van-Krevelen mechanism, where oxygen is in zero order while propane is in first order.60 However, in different catalytic systems, the types of active sites in vanadium-based catalysts might be varied, which include (1) bridged oxygen group (VO); (2) bridged oxygen between vanadium and support (V–O–S); and (3) bridged oxygen between adjacent vanadium atoms (V–O–V). Therefore, making clear the type of active sites in a specific catalytic system and proposing novel reaction mechanisms are the remaining tasks for the future in-depth investigation of vanadium-based catalysts.
In addition to the above controversial active centers, the design of the VOx-based catalyst also faces some challenges. In the O2-ODHP area, extensive studies have been made on VOx-based catalysts.61–63 It has been demonstrated that the catalytic behavior over VOx is related to a series of parameters such as the oxidation state, coordination number, aggregation state, and reducibility of vanadium species. These parameters are not only affected by the loading amount of V but also by the properties of the carrier and the preparation of the catalyst.64 Moreover, through the Mars–van-Krevelen mechanism, hydrocarbon molecules will react with oxygen species in VOx to generate alkene and H2O, and then reduced VOx should be re-oxidized by gaseous oxygen molecules to regenerate the active VOx sites. In this case, the whole process depends on the redox ability of VOx-based catalysts, which is also important for VOx catalyst design.
3.2 Molybdenum oxide-based catalysts
Similar to vanadium-based catalysts, molybdenum-based catalysts are also active in O2-ODHP, but the activity of molybdenum-based catalysts is slightly inferior to their vanadium-based counterparts. It has been reported that the performance of molybdenum-based catalysts in O2-ODHP is mainly affected by (1) the density of Mo on the catalyst surface; (2) the size of MoOx species; and (3) the electronic structure of MoOx species.O bonds since weaker MoO bonds were involved in rate-determining C–H bond activation steps requiring lattice-given oxygen atoms. At similar Mo surface densities, the catalysts predominantly containing ZrMo2O8/ZrO2 exhibited higher activities and lower initial propylene selectivity than those predominantly containing MoO3 species.65 This result indicated that the size of ZrMo2O8 domains increased with increasing Mo surface density without significant changes in the local structure or surface properties.
 | ||
| Fig. 2 (a) Dependence of the initial propylene formation rate on Cs–Mo/Zr catalysts with different Cs:Mo atomic ratio. (b) Comparison of dependence of the experimental and predicted initial propylene formation rate on Cs–Mo/Zr catalysts with different Cs:Mo atomic ratios. Reproduced with permission.66 Copyright 2000 Elsevier. (c) NH3-DRIFTS spectra of Al2O3, FeAl, 1Mo9FeAl, MoAl, and MoO3. (d) Signal of C3H7D during C3H8-D2-TPSR over FeAl, 1Mo9FeAl, and MoAl.46 Copyright 2023 Springer. (e) Differential charge density diagrams of the (010) facet Mo2N single crystal;67 (f) HS-LEIS spectra of the top layer of the porous Mo2N single crystal. (g) Potential energy profile for propane dehydrogenation on the Mo2N (100) surface (C in gray, N in blue, Mo in green, and H in white). Copyright 2021 Wiley. | ||
Gong et al. used two strategies including the coupling of surface acid catalysis and selective oxidation from lattice oxygen over MoO3–Fe2O3, simultaneously, to promote propylene production.46 They successfully introduced atomically dispersed Mo species on γ-Fe2O3 to create acid sites via the formation of MoO3. The coupling strategies achieved a stable catalytic performance with 49% propane conversion and 90% propylene selectivity for more than 300 cycles. HR-XPS spectra showed the enrichment of Mo, which was beneficial for propane activation, and the increasing density of medium acid sites. NH3-DRIFTS spectra suggested that the addition of Mo species caused both Lewis acid and Brønsted acid sites (Fig. 2c). The enrichment of Mo and modulation of acid properties accelerated the propane activation. Meanwhile, Mo also regulated the lattice oxygen activity, which led to selective oxidative dehydrogenation instead of over-oxidation in γ-Fe2O3 as confirmed by C3H8-D2-TPSR results (Fig. 2d).
Similarly, Xie's group designed the coordination conditions of Mo atoms over single-crystalline Mo2N and MoN monoliths for enhancing the non-oxidative dehydrogenation of propane to propylene.67 Through simulation, the authors predicted the electron structures, suggesting that the high-density Lewis acid sites were from the top-layer Mo with electron deficiency (Fig. 2e). Experimentally, HS-LEISS analysis further confirmed the atomic termination layer of Mo atoms for both Mo2N and MoN. The electron-deficient status was favorable to receive electrons in C–H, accelerating the reactant dissociation and activation (Fig. 2f). The NH3-TPD test clarified that the number of effective active sites (Lewis acid sites) on porous single crystals was higher compared to that on polycrystals. Taking Mo2N as an example, the authors further calculated the energy barrier for each reaction step (Fig. 2g). The results showed that high-density acid sites achieved the low activation energy with a reasonable potential barrier. Therefore, the porous single-crystalline Mo2N and MoN monoliths reached 11% propane conversion and ≈95–97% propylene selectivity under the mild conditions of 500 °C. The above works emphasized two facts: (i) catalysts containing exposed Mo active sites had excellent activity and tunable selectivity in O2-ODHP and (ii) compared to blindly increasing the number of exposed sites, reasonably designing the effective active sites on the catalyst surface could improve the catalytic activity and selectivity considerably.
Despite extensive studies, the performance of molybdenum-based catalysts in O2-ODHP is far from satisfactory, and even not comparable to their vanadium-based counterparts. In addition, molybdenum-based catalysts in O2-ODHP are structure-sensitive. Sometimes, molybdenum-based catalysts obtained by similar preparation methods might exhibit different catalytic performances and even reaction pathways in O2-ODHP.68 All these disadvantages make it a challenging task to study molybdenum-based catalysts in O2-ODHP.
3.3. Cobalt oxide-based catalysts
The variability of cobalt valence states enables it to function as a catalyst for O2-ODHP. Cobalt oxide, cobalt-containing layered double-oxides (LDO), and cobalt-containing metal–organic frameworks have been investigated. | ||
| Fig. 3 (a) Propane conversion and propylene selectivity, 0.3 g catalyst, T = 600 °C, WHSVC3H8 = 0.4 h−1; (b) propylene formation rate tested under similar initial propane conversion; WHSVC3H8 = 4.5, 3.125 and 0.4 h−1 for Co-acac@S-1, Co/S-1 and Co-edta@S-1; (c) the potential energy surface diagram.70 Copyright 2006 Royal Society of Chemistry. (d) XANES spectra of Co-SIM and Co-AIM + NU-1000 before and after activation, along with the data for cobalt reference samples. (e) Reaction coordinate for propane ODH. ΔH503K in kcal mol−1.71 Copyright 2016 American Chemical Society. (f) Schematic representation of the preparation of NU-1000-supported bimetallic catalysts. The promoter ions are anchored via SIM (pink) and Co ions are anchored via AIM (blue). (g) TOF values at 230 °C, as determined by varying the space velocity of propane.72 Copyright 2017 American Chemical Society. | ||
Cobalt-based catalysts exhibit high activities in O2-ODHP application, but they still suffer from two main shortages. Firstly, the cobalt-based catalysts generally have strong oxidation ability. This property makes it easy for propane overoxidation, which ultimately leads to low selectivity towards propylene. What's more, due to the variability of cobalt valence states, the stability of cobalt-based catalysts during the O2-ODHP process is poor, which remains an important concern.
3.4. Nickel oxide-based catalysts
Nickel oxide-based (NiO) catalysts have been widely studied in O2-ODHP due to their low cost and comparable catalytic performance. The performance of NiO is generally poor, and several approaches have been developed to improve its performance, including (1) constructing mesoporous NiO; (2) doping NiO with foreign elements; and (3) loading NiO onto a suitable support. | ||
| Fig. 4 (a) O2-TPD spectra of nano-NiO-sg and meso-NiO-300 samples.76 Copyright 2010 Elsevier. (b) Pore size distribution of mesoporous NiO support and Ni–Mo–O catalysts [(a) Mo/Ni = 0.05, (b) Mo/Ni = 0.1, (c) Mo/Ni = 0.2, (d) Mo/Ni = 0.3, (e) Mo/Ni = 0.4, (f) Mo/Ni = 0.5]. (c) SEM images and particle size distribution (inset) of mesoporous Ni–Mo–O catalysts: Mo/Ni = 0.05. (d) TEM images of mesoporous Ni–Mo–O catalysts: Mo/Ni = 0.5. (e) H2-TPR profiles of mesoporous NiO and Ni-Mo-O catalysts [(a) Mo/Ni = 0.05, (b) Mo/Ni = 0.1, (c) Mo/Ni = 0.2, (d) Mo/Ni = 0.3, (e) Mo/Ni = 0.4, (f) Mo/Ni = 0.5].79 Copyright 2019 Elsevier B.V. (f) Catalytic data obtained at 300 °C for Ti–Ni–O catalysts as a function of TiO2 content.80 Copyright 2005 Elsevier. (g) O2-TPD spectra of pure and Ce/Nb-doped NiO catalysts (200 mg sample). (h) NH3-TPD spectra of pure and Ce/Nb-doped NiO catalysts (200 mg sample).81 Copyright 2010 Springer. (i) Proposed reaction mechanism for the oxidative dehydrogenation of C3H8 to C3H6 by O2 in the presence of HCl over CeO2-based catalysts.82 Copyright 2018 American Chemical Society. | ||
Liu's group fabricated ordered mesoporous Ni–Mo–O catalysts via a two-step method involving a hard template and impregnation steps.79 The results of pore size distribution displayed the ordered mesoporous characteristic of Ni–Mo–O catalysts with a low ratio of Mo/Ni (Fig. 4b). Similarly, SEM images revealed a mesoporous structure when the concentration of Mo was low (Fig. 4c). TEM images suggested that the elimination of the mesoporous structure with the higher concentration of Mo should be attributed to the blockage of mesoporous channels due to the impregnation of Mo species (Fig. 4d). H2-TPR characterization showed that the addition of Mo could inhibit the reducibility of the Ni species according to the shift of the characteristic peaks toward the high temperature side (Fig. 4e). With an excellent mesoporous morphology and suitable redox ability, the Ni–Mo–O catalyst with a Mo/Ni ratio of 0.4 shows the highest activity with a propylene yield of 15.6%. The authors believed that the Ni–Mo–O catalyst was favorable for the C–C bond cleavage, which was the reason for the high selectivity of propylene. On the other hand, the decreased reducibility also contributed to the high selectivity because pure NiO with high redox ability and a lot of chemisorbed oxygen species would have strong reaction activity during the first step of OHDP.
Besides, multielement-doped NiO catalysts were also developed for O2-ODHP. For instance, Ce3NbNiO catalysts have been prepared by a sol–gel method.81 Compared to pristine NiO, Ce3NbNiO presents a higher propane conversion with higher propene selectivity at a relatively low temperature (250 °C). The enhancement effects of both Ce and Nb were investigated. O2-TPD measurements confirmed that Ce-doping increased the number of electrophilic oxygen species (Fig. 4g), which were mostly responsible for the deep oxidation reactions, so the propane conversion rate was largely improved. For the role of the Nb element, NH3-TPD proved the formation of weak acid sites (Fig. 4h). Since the higher strength and more acid sites over the catalyst surface could decrease the selectivity to olefin. The weak acid sites induced by Nb-doping should contribute to a decline in surface acidity and further improve the propene selectivity of the Ce3NbNiO catalyst.
Besides doping Ni-based catalysts, introducing Ni into other oxide catalysts is another elemental doping idea to improve the activity and selectivity. For instance, Ni doped CeO2 nanorod catalysts for O2-ODHP have been reported.86 Based on the periodic DFT calculations, the authors proposed a potential strategy to improve catalyst performance towards O2-ODHP. In detail, surface hydroxylation enhanced the propylene selectivity via inhibiting the formation of oxygenates and the formation of acetone. Compared to surface oxygen species, hydroxyls were more beneficial for the H abstractors, leading to an elementary mechanistic transformation in propene formation from MvK to LHHW. Thus, the Ni doping and surface hydroxylation (Lewis base addition) could be a reliable strategy for achieving selective O2-ODHP. Another example of Ni as a foreign doping element was reported by Farin et al.87 This work fabricated mesoporous β-NiMoO4 using Ni as the guest ion. Due to the coordination by the copolymer COO− functions, Ni cations were stable in the main β-phase. The presence of Ni species and the large specific surface from the mesoporous structure maintained a high activity level (propane conversion = 14.1%) and also reached high propylene selectivity (propene selectivity = 72%).
Another work about the NiO/CeO2 catalyst was recently reported by Wang's group.82 The authors investigated the relationship between catalytic behavior and exposed facets of CeO2 nanocrystals. The results showed that the [1 1 0] and [1 0 0] facets on the nanorod had the highest activity, and the nanocube enclosed by [1 0 0] facets possessed the most selectivity for propylene evolution. More importantly, the NiO modification achieved a single-pass propylene yield of 55% and outstanding stability with more than 100 h activity. The DFT and mechanistic studies suggested that the determining factors for enhanced activity and selectivity should be the surface oxygen vacancy and the surface chloride coverage (Fig. 4i). In detail, the peroxide species (O22−), which was formed by adsorption of O2 on surface oxygen vacancies, activated chloride and led to a radical-like active chlorine species. Further, the active chlorine species induced the dissociation of the C–H bond in propane, forming propylene as a major product.
3.5. Mixed transition metal oxide catalysts
In addition to the monometallic catalysts, some mixed TMO catalysts have also been studied in O2-ODHP, such as V–Mo mixed oxide,90 Nb–V mixed oxide,91 Mo–V–Te–Nb mixed oxide,92 and so on. Take the application of V–Mo mixed oxide in O2-ODHP as an example. Alasiri et al. prepared a series of V–Mo mixed oxide catalysts with varied Mo/V ratios (Mo/V = 1/1, 7/3, 8/2, and 9/1) using a modified citrate–nitrate auto-combustion method and applied the catalysts for O2-ODHP.90 In the Raman spectra of V–Mo mixed oxides (Fig. 5a), a peak at 785 cm−1 assigned to the vibration of V–O–Mo in a polymolybdovanadate species indicated that in all of the V–Mo mixed oxide catalysts, there was an interaction between the molybdenum and vanadium metal ions, which was effective for O2-ODHP. In ODHP, the catalyst with a Mo/V ratio of 9:1 gave the highest yield and selectivity of propylene, reaching a propylene yield of 4.8% and a propylene selectivity of ∼100.0% at 500 °C.
 | ||
| Fig. 5 (a) Raman spectroscopy of MoVOx-1–4.90 Copyright 2019 Wiley. (b) Normalized UV-Raman spectra (385 nm) of bare P25 and vanadia-loaded samples. The signal from the used CaF2 window is marked with an asterisk. (c) Proposed reaction mechanism for the ODH of propane over vanadia-loaded titania (P25) based on operando and transient spectroscopic analyses.93 Copyright 2023, American Chemical Society. (d) Diagram of primary reaction steps.19 Copyright 2009 Springer. (e) Ni K-edge XANES spectra and (f) Ni K-edge FT-EXAFS spectra of Ni1/CA catalysts and reference samples.107 Copyright 2023 Wiley. | ||
Recently, Hess et al. designed a VOx/TiO2 catalyst.93 Compared to conventional P25, which always caused carbon deposition during the O2-ODHP process, the active sites was located at VOx instead of TiO2. And UV-Raman spectroscopy showed that vanadia-related features at 858, 936, 991, and 1024 cm−1 were attributed to interface V–O–Ti, bridging V–O–V, the terminal vanadyl bond of V2O5, and the terminal vanadyl bond of amorphous vanadia, respectively (Fig. 5b). The O2-ODHP reaction involved preferentially the VO bonds of dimeric species instead of doubly bridged V–O–V bonds. With a high density of V active sites, the oligomeric V with better reducibility avoided the undesired oxidation process. The operando/transient spectroscopic results clarified that TiO2 could transfer H species from propane to V active sites based on Ti–OH groups on anatase (Fig. 5c). Therefore, the determining step of the C–H bond dissociation could be accelerated, improving the whole O2-ODHP efficiency.
As a typical p-type semiconductor, NiO can adsorb various types of oxygen species on its surface even under mild conditions.94 Thus, it has been confirmed that NiO is able to realize O2-ODHP at low temperatures, suggesting its great potential for the practical industry.95,96 However, there are still some challenges in NiO-based catalytic systems. NiO with poor stability is easily reduced to Ni0, and single NiO is hard to use as a stable catalyst with high catalytic performance.97 Although some NiO-based composite oxides, such as Ni–Ce–O, Ni–Nb–O, and Ni–Ti–O, have been developed for improving stability during the O2-ODHP process, the propylene yield rates are still very low.80,89,98 Therefore, how to simultaneously improve stability and reaction efficiency is an urgent problem when designing NiO-based catalysts. Recently, it has been reported that polyoxometalates possess the ability to activate molecular oxygen at moderate temperatures, so this new family has been developed for selective oxidation of light alkanes.99,100 Introducing polyoxometalates into NiO-based catalysts might be a reliable strategy to overcome the current problems.
4. Transition metal-based catalysts
Transition metal-based catalysts are generally applied in the direct dehydrogenation of propane. Hence, their studies in O2-ODHP are quite limited. The most common transition metal-based catalysts in O2-ODHP are Pt-based catalysts. Pt-based catalysts have been used for the total combustion of propane, which is the prevailing reaction at temperatures lower than 500 °C.101 In 2009, Vajda et al. reported that the subnanometer Pt cluster as the active phase exhibited excellent performance in O2-ODHP.19 Pt8–10 atomic clusters gave a propane conversion rate of 21.9% and a selectivity towards propylene of 64.0% at 500 °C, with the turnover frequency far superior to that of the classic VOx/Al2O3. Theoretical DFT calculations suggested that on the Pt clusters, compared to other C–H and C–C bonds, the C–H bond on the methylene group of propane was more likely to break first, forming propyl radicals, which then extract the protons from the methyl group to generate propylene (Fig. 5b). The excellent catalytic performance of Pt clusters might be attributed to the sub-nanometer level Pt8–10 active phase and its high dispersion. The main problem in Pt-based catalytic systems is agglomeration and sintering. To deal with this problem, Furukawa's group designed a Pt–Co–In ternary nanoalloy on a CeO2 support. The strong activation ability in the (Pt1−xCox)2In3 pseudo-binary alloy and the oxygen releasing ability in CeO2 facilitated Mars–van Krevelen-type coke combustion, thus improving the catalyst stability.34 In addition to transition metal-based catalysts containing noble metals, Yao et al. reported a cheap Ni-based catalyst (Ni@BOx/BN).102 With the assistance of the Ni subsurface, the in situ growth of BOx significantly increased the catalytic activity. The authors believed that the enhancement of catalytic efficiency should be attributed to the strong affinity of the Ni subsurface, which reduced the energy barrier for producing active species. In this case, the balance between B–OH cleavage and regeneration of boron hydroxyl groups could be achieved under low-temperature conditions.Besides the above metallic and alloyed transition metal-based catalysts, single-atom catalysts (SACs) are one of the most recent, revolutionary, and rapidly developing research fields in catalytic science.103 SACs inherit the advantages of easy separation and good recyclability of supported metal nanostructured catalysts and also combine the characteristics of homogeneous catalysts with highly homogeneous active centers and an adjustable coordination environment.104 The coordination between single atoms and the support involves strong interaction or charge transfer, conferring unique electronic and geometrical structures to individual metal atoms, differing from that of conventional metal NPs and carrying some charge.105 The high activity of valence electrons, the quantum confinement effect, and the sparse quantum level of metal atoms contribute to the maximum surface free energy of the metal species in SACs, which facilitates chemical interactions between single atoms and the support.105 In the O2-ODHP field, some SAC catalysts were also designed and fabricated. For example, Liu's group achieved the loading of PtSn-SA over commercial nano-Al2O3 with 0.1 wt% Pt.106 By combining in situ characterization and theoretical calculations, the Sn1Pt single-atom alloy structure could be confirmed, showing high efficiency with 47.6 mol gPt−1 h−1 of C3H6 evolution rate and more than 40 h of long-term stability. The results of in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFT) suggested that the coke deposition could be inhibited via Sn1Pt single-atom alloy mediated reconstruction, contributing to stability enhancement. Besides the noble-metal SACs, Zhang et al. reduced the size of transition metals and synthesized a Ni-based single-atom catalyst for O2-ODHP.107 X-ray absorption spectroscopy (XAS) confirmed the oxidation state (Ni3+) of Ni species on the CA support (Fig. 5c). EXAFS showed the isolated features of the metal Ni species, meaning the successful construction of the Ni-based SAC catalyst (Fig. 5d). Ni-SA supported on calcium aluminate was highly effective in O2-ODHP (Fig. 5e and f), recording a propylene selectivity of 47.3%, a yield of propylene of 14.2%, and good stability for 20 hours of testing, which was 2–3 times better than that of Ni nanoparticles. XPS characterization elucidated the reason for the improved efficiency and selectivity. The more positive electronic properties of the Ni-SA sites were beneficial for the activation of oxygen molecules (propylene desorption), effectively inhibiting further oxidation of propylene to COx by-products.
Although the above works proved that transition metal-based catalysts are efficient for O2-ODHP, the synthesis process of the clusters and single-atom catalysts is quite complex, requiring accurate and strict control, which limits the industrial application of clusters and single-atom catalysts in O2-ODHP. Meanwhile, in some transition metal-based catalysts containing noble metals, their high cost is also another disadvantageous factor, limiting their large-scale applications and leading to higher requirements in their activity, stability, and recyclability.
5. Rare earth metal oxide catalysts
Among the rare earth metal oxide catalysts for O2-ODHP, CeO2-based catalysts are mainly focused on because of their excellent oxygen storage and release properties,108,109 and strong redox properties.82,110The performance of pure CeO2 is still not ideal for industrialization, evidenced by the high propane conversion rate but extremely low propylene selectivity, owing to the occurrence of overoxidation. Modifying CeO2 by NiO,82 Cl,110 F,111 or P112 could improve its performance. For instance, Wan et al. demonstrated that F-modified CeO2 could selectively promote the production of propylene.111 After further modification by an appropriate amount of Cs, the catalyst with the composition 3% Cs2O/CeO2–2CeF3 achieved a propylene yield of more than 30.0% at 500 °C. CeF3 played two functions in 3% Cs2O/CeO2–2CeF3 for O2-ODHP: one is that the surface F replaced some lattice oxygen in CeO2, promoting the migration of oxygen species and the activation of surface oxygen species; and another is that the addition of F resulted in a highly dispersed surface CeO2 active phase. The modification of CeO2 by P could also facilitate the dispersion of the surface active phase CeO2, thus improving propylene selectivity.112 As a result, at the reaction temperature of 550 °C, CePO4 recorded a propylene selectivity of 62.0%, much higher than that of CeO2 (31.0% under the same reaction conditions).
Although there are some studies on the application of rare earth CeO2-based catalysts in O2-ODHP, the enhancement effect from the rare earth metal oxides has not been extensively investigated and utilized in the O2-ODHP area. The reaction mechanism is not clear yet. Thus, more efforts should be devoted to this kind of catalytic system to elucidate the possible theories.
6. Non-metallic catalysts
Non-metallic catalysts, such as boron nitride (BN), other B-containing catalysts, and C-based catalysts, have also been developed for O2-ODHP. In this sub-section, we will review the current progress of these catalysts in O2-ODHP.6.1. BN catalysts in O2-ODHP
Among the non-metallic catalysts, the performance of BN is relatively excellent,113 Since there have been some reviews on the development of BN catalysts,114–117 the progress of BN in only O2-ODHP is briefly summarized here.The application of BN in O2-ODHP was initiated in 2016 when the Hermans group reported that commercial hexagonal BN and BN nanotubes exhibited much higher catalytic activity and propylene selectivity in O2-ODHP than other catalysts.118 For example, on a commercial hexagonal BN catalyst, at a reaction temperature of 490 °C, the conversion of propane was 14.0%, the selectivity of propylene could reach 79.0%, and there was almost no complete oxidation product CO2 generated. In addition, BN also exhibited very high stability in O2-ODHP. The rich hydroxyl groups at the edge endowed BN with an initial propane conversion rate of 20.6% and an initial propylene selectivity of 80.2% at a reaction temperature of 530 °C in O2-ODHP.119 Besides, its activity and selectivity remained almost unchanged within 300 hours. The excellent catalytic performance of BN has opened up a new path for the selective cleavage of C–H bonds in alkanes and has become a research hotspot. From then on, several approaches have been adopted to improve the performance of BN in O2-ODHP, which include (1) increasing the specific surface area of BN;120 (2) doping BN with foreign elements;121 (3) preparing BN with a local chemical environment regulated by plasma, and so on.122 Here some typical examples are illustrated.
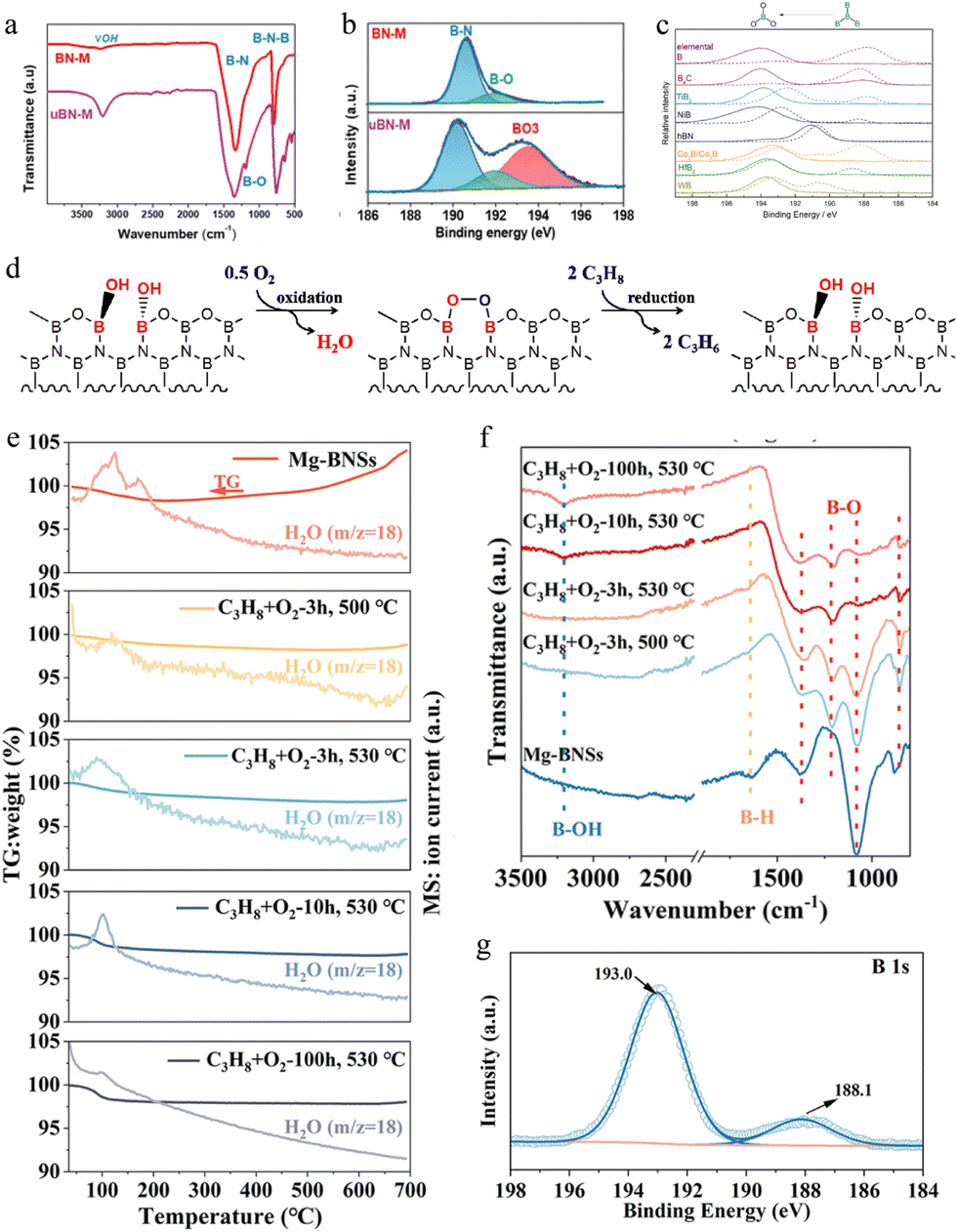 | ||
| Fig. 6 (a) FTIR and (b) B 1s high-resolution spectra of BN-M and uBN-M.124 Copyright 2021 American Chemical Society. (c) The XPS B 1s feature of fresh (dotted) and spent (solid) B-containing catalysts.123 Copyright 2016 Science. (d) The proposed redox reaction cycle in the oxidative dehydrogenation of propane over boron nitride.126 Copyright 2018 Elsevier. (e) TG-MS analysis of Mg-BNSs and their spent forms under an O2-rich atmosphere (20% O2 + 80% N2). (f) FT-IR spectra of Mg-BNSs and the spent Mg-BNSs activated in the reaction atmosphere at different temperatures and durations. (g) B 1s XPS spectrum of Mg-BNSs tested for 100 h over ODHP.18 Copyright 2023 Royal Society of Chemistry. | ||
In order to study the active sites of the BN catalyst in O2-ODHP, understand the reaction pathway of O2-ODHP on the BN catalyst at the molecular/atomic level, and lay a theoretical foundation for the design of an efficient BN catalyst, researchers analyzed the structure of the BN catalyst and explored the mechanism of BN in O2-ODHP. For example, combining kinetic analysis and theoretical calculations, Hermans et al. proposed that the oxygen terminal site on the hexagonal BN armchair edge ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) B–O–O–B
B–O–O–B![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) was the catalytic active site, B–O–O–B activated the second hydrogen atom of propane, while breaking the O–O bond to form boron hydroxyl groups and nitrogen oxygen radicals.118 The authors further analyzed the surface elements of the catalyst using XPS and found that in O2-ODHP, BOx species gradually formed on the BN surface and tended to stabilize, suggesting that the BOx species were the active site.125 Hermans et al. used 1H–11B coupling nuclear magnetic resonance and X-ray absorption spectroscopy to investigate the structural changes of the BN catalyst before and after the reaction, determining that the active site of the BN catalyst was a three coordinated boron site (B(OH)xO3−x) with a variable number of hydroxyl groups and bridging oxidation groups, rather than the oxygen terminal site on the armchair edge B–O–O–B.123 Lu et al. found that BN exhibited excellent activity and olefin selectivity only after undergoing an activity induction period in O2-ODHP. XPS and in situ XRD characterization confirmed that surface BOx species gradually formed and tended to stabilize during the oxidative dehydrogenation reaction (Fig. 6c), showing a consistent trend with catalyst activity, confirming that BOx species were active sites as proposed by Hermans et al.126,127 However, further combining kinetic analysis, Lu et al. claimed that the B–OH dynamically generated on the catalyst surface was the active site of the catalyst (Fig. 6d).
was the catalytic active site, B–O–O–B activated the second hydrogen atom of propane, while breaking the O–O bond to form boron hydroxyl groups and nitrogen oxygen radicals.118 The authors further analyzed the surface elements of the catalyst using XPS and found that in O2-ODHP, BOx species gradually formed on the BN surface and tended to stabilize, suggesting that the BOx species were the active site.125 Hermans et al. used 1H–11B coupling nuclear magnetic resonance and X-ray absorption spectroscopy to investigate the structural changes of the BN catalyst before and after the reaction, determining that the active site of the BN catalyst was a three coordinated boron site (B(OH)xO3−x) with a variable number of hydroxyl groups and bridging oxidation groups, rather than the oxygen terminal site on the armchair edge B–O–O–B.123 Lu et al. found that BN exhibited excellent activity and olefin selectivity only after undergoing an activity induction period in O2-ODHP. XPS and in situ XRD characterization confirmed that surface BOx species gradually formed and tended to stabilize during the oxidative dehydrogenation reaction (Fig. 6c), showing a consistent trend with catalyst activity, confirming that BOx species were active sites as proposed by Hermans et al.126,127 However, further combining kinetic analysis, Lu et al. claimed that the B–OH dynamically generated on the catalyst surface was the active site of the catalyst (Fig. 6d).
Besides the above-mentioned several species, boron atoms at the defective sites,128 boron-atom-terminated zigzag,129 and N2O or NOx-type sites130 have also been reported as active sites on BN in O2-ODHP. Therefore, currently, there is still controversy over the active site of BN in O2-ODHP. Researchers need to devote more effects to make clear which one is the intrinsic active site for O2-ODHP over a specific catalytic system.
6.2. Other B-containing catalysts
In addition to BN, some other B-containing catalysts, such as exfoliated boron nanosheets,18 boron carbonitride,131 boron phosphate (BPO4),132 boron carbide,133 boron phosphide and silicon boride,134 and boron-containing metal–organic frameworks,7 have also been developed for catalytic O2-ODHP. Here we take the application of exfoliated boron nanosheets in O2-ODHP as an example for illustration. Zhang et al. exfoliated layered MgB2 with hydrochloric acid to synthesize exfoliated boron nanosheets (Mg-BNSs) for O2-ODHP.18 It was revealed that the exfoliated boron nanosheet afforded a propylene and ethylene selectivity of 63.5% and 18.4%, respectively, at a propane conversion of 40.0% at 530 °C, with the olefin productivity much superior to that of the commercial h-BN and other reported boron-based catalysts. In addition, the exfoliated boron nanosheet maintained excellent stability in O2-ODHP at 530 °C for 100 hours. The dynamic evolution of their structure and surface functional groups under reaction conditions have been investigated for clarifying the origin of the excellent performance of Mg-BNSs. Thermogravimetric analysis with mass spectroscopy (TG-MS) showed an obvious weight loss of fresh Mg-BNSs under 200 °C (Fig. 6e). However, the mass increase started at 400 °C, suggesting the continuous oxidation of Mg-BNSs under an oxygen-rich atmosphere. In contrast, if the sample was activated at 500 or 530 °C for over 3 h under the reaction atmosphere, almost no mass decline could be monitored. Thus, the oxidized form was the main factor for the high activity, because it could avoid the coke formation. In addition, the FTIR spectra observed the B–OH and B–O bonds at 1367, 1200, and 860 cm−1 (Fig. 6f), which have been reported as the real active sites for O2-ODHP. The spent Mg-BNSs exposed more oxygen atoms at the surface and exhibited a more pronounced peak for B–O in the B 1s XPS spectrum (Fig. 6g). These results indicated that the activity of Mg-BNSs was from a large number of generated B–O active sites. This work displayed the huge potential of B-based catalysts for O2-ODHP under harsh reaction conditions. Considering the low price of MgB2, B-based catalysts could be a better choice for the O2-ODHP reaction.6.3. C-Based catalysts
In 1987, Schraut et al. unexpectedly discovered that carbon deposition could induce the oxidative dehydrogenation reaction of ethylbenzene, which opened up the research on C-based catalysts in the field of oxidative dehydrogenation of low-carbon alkanes. Nanocarbon is a promising catalyst for O2-ODHP due to its abundant reactive oxygen species, resistance to carbon deposition, and unique electronic and structural properties. However, the unsatisfactory propylene selectivity and low durability under high temperature or O2-rich conditions restrict the development of carbon-based catalysts. Fortunately, more and more strategies have been proposed to overcome these disadvantages and improve the catalytic performance of nanocarbon catalysts toward O2-ODHP implementation.Different C-based catalysts with 2D or 3D morphologies, including carbon nanotubes (CNTs),135 carbon nanofibers,136 nanodiamonds,137etc., have been applied in O2-ODHP. Generally, these C-based catalysts have been modified with the aim of improving their catalytic performance. For instance, Frank et al. modified carbon nanotubes with boron or phosphorus and investigated the effects of heteroatoms in O2-ODHP.135 It was revealed that the introduction of both boron and phosphorus increased the apparent activation energy of the catalyst and the reaction order of oxygen while affecting little the reaction order of propane. It indicated that boron or phosphorus can inhibit the formation of electrophilic oxygen-active sites, where phosphorus was more effective. The inhibited formation of electrophilic oxygen active sites prohibited the overoxidation of propane and propylene and thus improved the selectivity towards propylene. As a result, the modification of carbon nanotubes with phosphorus enhanced the thermal stability of carbon nanotubes, achieving a propane conversion of 7.0% at a selectivity of propylene of about 50.0% (Fig. 7a and b). Chen et al. doped nitrogen into carbon nanotubes to regulate the types of functional groups on their surface, and investigated their performance in O2-ODHP.138 It was demonstrated that nitrogen doping helped to increase the surface charge density of carbon nanotubes, leading to a strengthened repulsive force between carbon nanotubes and propylene. It was beneficial for reducing the adsorption of propylene on the catalyst, avoiding the overoxidation of propylene, and consequently improving the selectivity towards propylene (Fig. 7c and d).
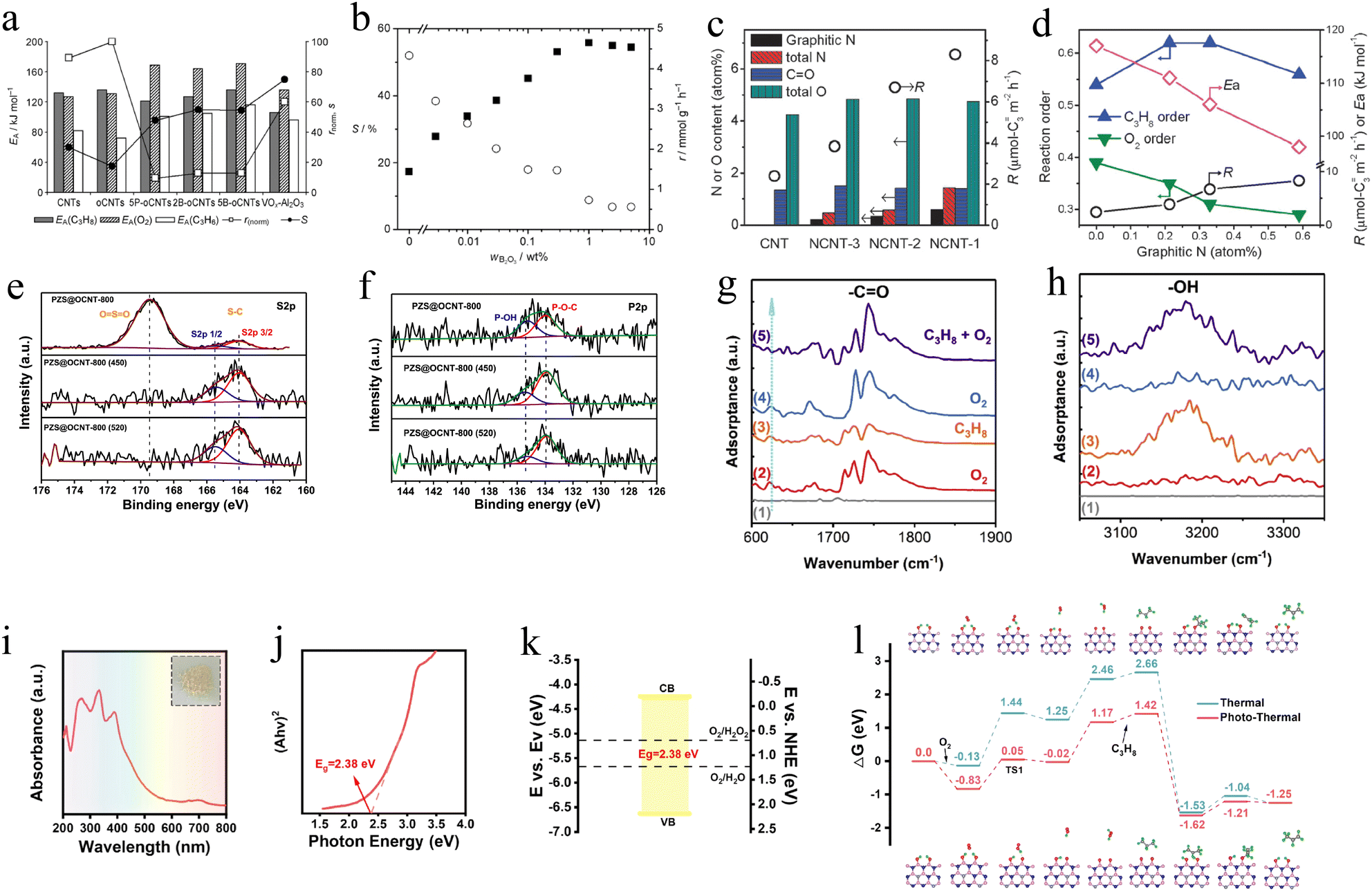 | ||
| Fig. 7 (a) Catalytic performance of carbon nanotube catalysts in ODHP, with VOx–Al2O3 as a reference. (b) Propylene selectivity at 5% propane conversion (■) and reaction rate r (○) as a function of B2O3 loading (WB2O3). Reproduced with permission from ref. 135. Copyright 2009 Wiley. (c) Structural parameters and C3H6 formation rates of spent catalysts. (d) Reaction orders, C3H6 formation rates, and activation energy as a function of graphitic N content. Reproduced with permission.138 Copyright 2013 Royal Society of Chemistry. Deconvolution of (e) S 2p and (f) P 2p XPS signals for PZS@OCNT-800 before and after ODHP reactions (450 °C and 520 °C).139 Copyright 2022 Elsevier. (g) and (h) In situ DRIFT difference spectra at steady state after switching various reaction atmospheres at 500 °C.140 Copyright 2020 Elsevier. (i) UV-DRS of SS-BCNNSs. (j) Dependence of (Ahv)2 on the photon energy. (k) Schematic band structure of SS-BCNNSs. (l) DFT calculated energy profile for ODHP under thermal and photo-thermal conditions.141 Copyright 2023 Elsevier. | ||
Another strategy was reported by Qi et al., who utilized heteroatom co-doped polymers (PZS) and oxidized CNTs, achieving 63% propylene selectivity at 14.3% propane conversion.139 What's more, the poor stability of CNTs has also been improved, realizing more than 20 h O2-ODHP reactions at 520 °C. The kinetic tests conducted at different propane and oxygen partial pressures demonstrated that the as-prepared CNT-based catalysts have excellent resistance to oxidation and carbon deposition, which is the reason for the enhanced stability. The XPS spectra could detect the P–O and S–C species on the catalyst surface, which contributed to the improvement of propylene selectivity and oxidation resistance (Fig. 7e and f).
In spite of the fact that C-based catalysts exhibited a certain activity in O2-ODHP, due to the complex types of functional groups on the surface of C-based materials (such as hydroxyl group, carbonyl group, anhydride group, lactone group, etc.), the qualitative and quantitative determination of effective functional groups has always been a research difficulty in the field of O2-ODHP.142 In addition, the complex variety of functional groups on the surface of C-based catalysts makes the reaction more complex. The oxidative dehydrogenation process as well as the overoxidation of propane and propylene might occur. The multiple parallel reaction pathways and diverse intermediates make it challenging to use C-based catalysts in O2-ODHP.143
A series of carbon nitride (CN) polymers provided more opportunities for further developing C-based catalysts with high selectivity in the O2-ODHP field. For instance, graphitic CN has been reported as an O2-ODHP catalyst achieving a propane conversion of 12.8% and a selectivity of 74.7% for the desired propylene product.140In situ DRIFTS characterization showed the orderly formation of –CO and –OH species (Fig. 7g and h). Accordingly, the authors speculated the reaction pathway. Specifically, the CO abstracted H atoms from propane to generate propylene and formed C–OH. Subsequently, the following O2 treatment extracted H atoms from C–OH species and re-generated CO groups. The switch between CO and C–OH groups under the reactant atmosphere drove the O2-ODHP process while avoiding the direct interaction between C3H8 and O2. Theoretically, DFT calculations predicted a reasonable O2-ODHP reaction pathway, during which two adjacent CO groups on the edge of CN can activate two C–H bonds of propane simultaneously, thus avoiding the generation of free C3H7* radicals and leading to the high selectivity of light olefins.
Due to the high light absorption ability of C-based materials, photothermal catalytic technology has emerged as a novel strategy to trigger O2-ODHP reaction under mild conditions. Compared to traditional reaction conditions, the lower reaction temperature during photothermal catalysis benefits the control of propylene selectivity and catalyst stability. One recent example is based on a semiconducting boron CN (BCN) catalyst, which integrated photocatalysis with thermal catalysis.141 The fundamental theory is the cooperation between electrically conductive properties from the graphene phase and insulation from boron nitride (h-BN). Thus, the BCN alloy could possess both excellent catalytic performance of h-BN towards O2-ODHP and the semiconductor behavior of graphene. Ultraviolet-visible diffuse reflectance spectroscopy (UV-DRS) showed the effective visible light response of BCN with a 500 nm absorption edge (Fig. 7i). And the Tauc plot clarified that BCN was a direct band gap material, which had a band gap of 2.83 eV (Fig. 7j). According to ultraviolet photoelectron spectroscopy (UPS) measurements, the authors determined the energy band structure of BCN with 3.92 eV vs. evac conduction band and 6.62 eV vs. evac valence band, indicating the photocatalytic ability for activating O2 (Fig. 7k). At 480 °C, the propane conversion increased from 11.1% to 21.2% via light irradiation, resulting in an increase in the overall yield of olefin from 10.4% to 19.6% and higher propylene selectivity. DFT results clearly showed the advantage of photothermal catalysis in terms of the energy barrier and the rate-determining step (the dehydrogenation of B-OH groups to form BO·) in the BCN catalytic system (Fig. 7l). Indeed, this work confirmed the great potential of integrating photocatalysis with conventional thermal catalysis via semiconductor catalysts to enhance propane conversion and propylene selectivity during the O2-ODHP reaction. The photothermal method will play a key role in achieving carbon neutrality.
7. Summary and outlook
To date, a wide variety of catalysts have been developed for O2-ODHP, such as transition metal oxide catalysts, transition metal-based catalysts, rare earth metal oxide catalysts, and non-metallic catalysts. Some typical examples of the catalysts are displayed in Table 1. The current challenges and coping strategies are summarized below.| Catalyst | Reaction condition | Performance | Ref. |
|---|---|---|---|
| V2O5/SiO2 | Reactor type: conventional continuous flow quartz microreactor | Selectivity towards propylene is 72.0% | 16 |
| Reactant ratio: C3H8:O2:N2:He = 2:1: 1:6 |
|||
| Reactant feed: GHSV = 1700 h−1 | |||
| Reaction temperature: 450 °C | |||
| Reaction pressure: 1 atm | |||
| ZrMo2O8/ZrO2 | Reactor type: packed-bed tubular quartz reactor | Selectivity towards propylene is 80.0% | 23 |
| Reactants: propane and oxygen at 14.03 and 1.74 kPa | |||
| Reaction temperature: 500 °C | |||
| Ti–Ni–O | Reactant ratio: C3H8:O2:N2 = 1.1:1:4 |
Propane conversion is about 28.4%, selectivity to propylene is 42.5% and propylene yield is 12.1% | 36 |
| Reactant feed: 9000 mL h−1 g−1 | |||
| Reaction temperature: 300 °C | |||
| MoVOx | Reactor type: fixed bed reactor | Propane conversion is about 2.7%, selectivity to propylene is 100% and propylene yield is 2.7% | 45 |
| Reactant ratio: C3H8:O2:N2 = 6:3:91 |
|||
| Reaction temperature: 350 °C | |||
| Pt–Ce/γ-Al2O3 | Reactor type: freestanding anaerobic-anoxic-oxic flow reactor setup | Propane conversion is about 37.0% and selectivity to propylene is 96.0% | 49 |
| Reactant ratio: C3H8:H2:Ar = 1:1:5 |
|||
| Reaction temperature: 576 °C | |||
| Ni-CA SACs | Reactor type: fixed bed reactor | Selectivity to propylene is 47.3% and propylene yield is 14.2% | 53 |
| Reactant ratio: C3H8:O2:N2 = 2:1:17 |
|||
| Reactant feed: WHSV = 12000 L kgcat−1 h−1 |
|||
| Reaction temperature: 580 °C | |||
| Reaction pressure: 1 atm | |||
| NiO−CeO2 | Reactor type: fixed-bed flow reactor | Propane conversion is about 69.0%, selectivity to propylene is 80.0% and propylene yield is 55.0% | 57 |
| Reactant ratio: C3H8:HCl:O2 = 9:9:5 |
|||
| Reactant feed: 48 mL min−1 | |||
| Reaction temperature: 500 °C | |||
| Reaction pressure: 1 atm | |||
| P-CeO2 | Reactor type: fixed-bed quartz tube down-flow reactor | Propane conversion is about 8.7% and selectivity to propylene is 70.1% | 59 |
| Reactant ratio: C3H8:air = 1:2.5 |
|||
| Reactant feed: VHSV = 9000 h−1 | |||
| Reaction temperature: 600 °C | |||
| h-BN | Reactor type: quartz tube reactor | Propane conversion is about 14.0% and selectivity to propylene is 79.0% | 65 |
| Reactants: 0.15 atm O2 and 0.3 atm C3H8 | |||
| Reaction temperature: 490 °C. | |||
| BNOH | Reactor type: packed-bed quartz microreactor | Propane conversion is about 20.6% and selectivity to propylene is 80.2%. | 66 |
| Reactant ratio: C3H8:O2:He = 1:1.5:3.5 |
|||
| Reactant feed: 192 mL min−1 | |||
| Reaction temperature: 530 °C | |||
| BN with a larger specific surface area | Reactor type: fixed-bed reactor | Propane conversion is about 50.0% and selectivity to propylene is 70.0% | 67 |
| Reactant ratio: C3H8:O2:He = 1:50:49 |
|||
| Reactant feed: 20 mL min−1 | |||
| Reaction temperature: 525 °C | |||
| Reaction pressure: 1 atm | |||
| N2 plasma treated BN | Reactor type: packed-bed quartz reactor | Propane conversion is 26.0% and the total selectivity towards propylene and ethene is 89.4% | 69 |
| Reactant ratio: C3H8:O2:Ne = 1:1.5:3.5 |
|||
| Reactant feed: 48 mL min−1 | |||
| Reaction temperature: 520 °C | |||
| Reaction pressure: atmospheric pressure | |||
| B4C | Reactor type: quartz reactor tube | Propane conversion is about 5.1% and selectivity to propylene is 86.0% | 73 |
| Reactant ratio: C3H8:O2:N2 = 6:3:11 |
|||
| Reactant feed: WHSV−1 = 10.5 kg-cat * s mol−1 C3H8 | |||
| Reaction temperature: 500 °C | |||
| Reaction pressure: 0.30 atm C3H8 and 0.15 atm O2 | |||
| Ti2B | Reactor type: quartz reactor tube | Propane conversion is about 5.2% and selectivity to propylene is 85.3% | 73 |
| Reactant ratio: C3H8:O2:N2 = 6:3:11. |
|||
| Reactant feed: WHSV−1 = 12.5 kg-cat * s mol−1 C3H8 | |||
| Reaction temperature: 500 °C | |||
| Reaction pressure: 0.30 atm C3H8 and 0.15 atm O2 | |||
| Exfoliated layered MgB2 | Reactor type: fixed-bed reactor | Propane conversion is about 39.8%, selectivity to propylene is 63.5% and propylene yield is 26.3% | 78 |
| Reactant ratio: C3H8:O2:N2 = 1:1:3 |
|||
| Reactant feed: WHSV = 24000 mL g−1 h−1 |
|||
| Reaction temperature: 530 °C | |||
| Reaction pressure: atmospheric pressure | |||
| N doped CNT | Reactor type: immobilized bed quartz reactor | Propane conversion is about 5.0% and selectivity to propylene is 56.5% | 82 |
| Reactant ratio: C3H8:O2: He = 2:1:19 |
|||
| Reactant feed: GHSV = 13 L g−1 h−1 | |||
| Reaction temperature: 400 °C | |||
| Reaction pressure: atmospheric pressure |
(1) Despite the variety of catalysts developed for O2-ODHP, to the best of our knowledge, the highest yield towards propylene was achieved over the NiO–CeO2 catalyst, up to 55.0%, with a propane conversion of 69.0% and a selectivity towards propylene of 80.0%. Apparently, their performances are still far away from industrialization. In addition, for most of the catalytic systems, it is still challenging to realize a high propane conversion rate and high selectivity towards propylene simultaneously. Therefore, in the future, it is demanded to develop a novel catalyst, which can achieve high selectivity towards propylene at high propane conversion rates.144,145
(2) Even though characterization and theoretical calculations have been conducted over some of the catalytic systems, the intrinsic mechanism remains unclear. As is known, the catalytic reaction mechanism plays an important role in guiding the development of more advanced and efficient catalysts. Therefore, in the future, more studies should be focused on the investigation of the reaction system, using in situ/operando characterization techniques as well as theoretical simulations, to unravel the intrinsic active site and reaction pathways of the O2-ODHP process.
(3) Considering the two facts that (i) propane as the reactant has to be extracted from other raw materials like shale gas and (ii) propylene is important for downstream chemical reactions to get high value-added products, the coupling between O2-ODHP and other catalytic reactions should be paid more attention to realize the large-scale industrialization and even achieve the green chemistry and carbon neutral aims. For this aim, a cheaper catalyst should be designed and developed to meet the requirements of continuous and large-scale catalytic reactions. What's more, the reaction vessel for conducting different catalytic processes should also be constructed to promote the advanced catalytic systems involving the combination of the shale gas–propane conversion and O2-ODHP, and the coupling between O2-ODHP and reactions converting propylene into value-added products.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received financial support from the Young Talent Plan of Liaoning Province (XLYC2203068), Scientific Research Foundation of Technology Department of Liaoning Province of China (2022-MS-379), and National Natural Science Foundation of China (21902116). Financial support from the program of China Scholarships Council (No. 202206250016) is gratefully acknowledged.Notes and references
- W. Wu, W. Guo, W. Xiao and M. Luo, Chem. Eng. Sci., 2011, 66, 4722–4732 CrossRef CAS.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 1546–1548 CrossRef CAS PubMed.
- H. Oikawa, Y. Shibata, K. Inazu, Y. Iwase, K. Murai, S. Hyodo, G. Kobayashi and T. Baba, Appl. Catal., A, 2006, 312, 181–185 CrossRef CAS.
- L. Schumacher, J. Shen, K. Hofmann and C. Hess, Catal. Today, 2024, 426, 114387 CrossRef CAS.
- M. L. Sarazen and C. W. Jones, J. Phys. Chem. C, 2018, 122, 28637–28644 CrossRef CAS.
- X. Fan, D. Liu, X. Sun, X. Yu, D. Li, Y. Yang, H. Liu, J. Diao, Z. Xie, L. Kong, X. Xiao and Z. Zhao, J. Catal., 2020, 389, 450–460 CrossRef CAS.
- L. Cao, P. Dai, S. Wen, Y. Jiang, D. Liu, X. Gu, Q. Zhang, Y. Xia, G. Zhong, X. Zhao and J. Xie, Matter, 2023, 6, 4376–4387 CrossRef CAS.
- A. Al-Douri, D. Sengupta and M. M. El-Halwagi, J. Nat. Gas Sci. Eng., 2017, 45, 436–455 CrossRef CAS.
- J. Guo, Y. Lei, H. Liu, Y. Li, D. Li and D. He, Catal. Sci. Technol., 2023, 13, 4045–4063 RSC.
- Y. Li, Y. Lei, D. Li, A. Liu, Z. Zheng, H. Liu, J. Guo, S. Liu, C. Hao and D. He, ACS Catal., 2023, 10177–10204 CrossRef CAS.
- Y. Lei, J. Ye, J. García-Antón and H. Liu, Chin. J. Catal., 2023, 53, 72–101 CrossRef CAS.
- S. Hu, P. Qiao, X. Yi, Y. Lei, H. Hu, J. Ye and D. Wang, Angew. Chemie, 2023, 135, e202304585 CrossRef.
- Y. Lei, Z. Jia, H. Hu, L. Liu, J. Ye and D. Wang, Catalysts, 2022, 12, 1323 CrossRef CAS.
- S. Sokolov, V. Y. Bychkov, M. Stoyanova, U. Rodemerck, U. Bentrup, D. Linke, Y. P. Tyulenin, V. N. Korchak and E. V. Kondratenko, ChemCatChem, 2015, 7, 1691–1700 CrossRef CAS.
- S. Sokolov, M. Stoyanova, U. Rodemerck, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2014, 4, 1323 RSC.
- B. Hu, W. Kim, T. P. Sulmonetti, M. L. Sarazen, S. Tan, J. So, Y. Liu, R. S. Dixit, S. Nair and C. W. Jones, ChemCatChem, 2017, 9, 3330–3337 CrossRef CAS.
- A. Siahvashi, D. Chesterfield and A. A. Adesina, Ind. Eng. Chem. Res., 2013, 52, 4017–4026 CrossRef CAS.
- D. Zhang, S. Wang, C. Zhang, L. He and W. Sun, Nanoscale, 2024, 16, 1312–1319 RSC.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- T. Blasco and J. M. L. Nieto, Appl. Catal., A, 1997, 157, 117–142 CrossRef CAS.
- R. Grabowski, Catal. Rev., 2006, 48, 199–268 CrossRef CAS.
- P. Concepción, J. M. López Nieto and J. Pérez-Pariente, Catal. Lett., 1993, 19, 333–337 CrossRef.
- J. M. López Nieto, P. Concepción, A. Dejoz, H. Knözinger, F. Melo and M. I. Vázquez, J. Catal., 2000, 189, 147–157 CrossRef.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- Y. Yuan, W. N. Porter and J. G. Chen, Trends Chem., 2023, 5, 840–852 CrossRef CAS.
- J. H. Carter, T. Bere, J. R. Pitchers, D. G. Hewes, B. D. Vandegehuchte, C. J. Kiely, S. H. Taylor and G. J. Hutchings, Green Chem., 2021, 23, 9747–9799 RSC.
- Y. Gambo, S. Adamu, A. A. Abdulrasheed, R. A. Lucky, M. S. Ba-Shammakh and M. M. Hossain, Appl. Catal., A, 2021, 609, 117914 CrossRef CAS.
- X. Jiang, L. Sharma, V. Fung, S. J. Park, C. W. Jones, B. G. Sumpter, J. Baltrusaitis and Z. Wu, ACS Catal., 2021, 11, 2182–2234 CrossRef CAS.
- X. Gao, M. Liu, Y. Huang, W. Xu, X. Zhou and S. Yao, ACS Catal., 2023, 13, 9667–9687 CrossRef CAS.
- Z.-P. Hu, D. Yang, Z. Wang and Z.-Y. Yuan, Chin. J. Catal., 2019, 40, 1233–1254 CrossRef CAS.
- M. A. Atanga, F. Rezaei, A. Jawad, M. Fitch and A. A. Rownaghi, Appl. Catal., B, 2018, 220, 429–445 CrossRef CAS.
- E. Gomez, B. Yan, S. Kattel and J. G. Chen, Nat. Rev. Chem., 2019, 3, 638–649 CrossRef CAS.
- F. Xing, Y. Nakaya, S. Yasumura, K. Shimizu and S. Furukawa, Nat. Catal., 2022, 5, 55–65 CrossRef CAS.
- S. Song, X. Chen, Y. Fo, M. Yang, H. Su, K. Yang, X. Ji, X. Lv, Z. Li, Y. Wei, G. Huang, C. Xu, J. Liu and W. Song, Chem. Catal., 2023, 3, 100663 CrossRef CAS.
- E. Gomez, Z. Xie and J. G. Chen, AIChE J., 2019, 65, e16670 CrossRef.
- E. Gomez, S. Kattel, B. Yan, S. Yao, P. Liu and J. G. Chen, Nat. Commun., 2018, 9, 1398 CrossRef PubMed.
- L. M. Neal, S. Yusuf, J. A. Sofranko and F. Li, Energy Technol., 2016, 4, 1200–1208 CrossRef CAS.
- T. Wu, Q. Yu, K. Wang and M. van Sint Annaland, Catalysts, 2021, 11, 119 CrossRef CAS.
- S. Yusuf, V. Haribal, D. Jackson, L. Neal and F. Li, Appl. Catal., B, 2019, 257, 117885 CrossRef CAS.
- J. Baek, H. J. Yun, D. Yun, Y. Choi and J. Yi, ACS Catal., 2012, 2, 1893–1903 CrossRef CAS.
- S. Wang and Z. H. Zhu, Energy Fuels, 2004, 18, 1126–1139 CrossRef CAS.
- T. Wu, Q. Yu and Q. Qin, Pet. Sci. Technol., 2018, 36, 266–272 CrossRef CAS.
- E. A. de Graaf, G. Rothenberg, P. J. Kooyman, A. Andreini and A. Bliek, Appl. Catal., A, 2005, 278, 187–194 CrossRef CAS.
- N. Ballarini, F. Cavani, A. Cericola, C. Cortelli, M. Ferrari, F. Trifirò, G. Capannelli, A. Comite, R. Catani and U. Cornaro, Catal. Today, 2004, 91–92, 99–104 CrossRef CAS.
- X. Wang, C. Pei, Z.-J. Zhao, S. Chen, X. Li, J. Sun, H. Song, G. Sun, W. Wang, X. Chang, X. Zhang and J. Gong, Nat. Commun., 2023, 14, 2039 CrossRef CAS PubMed.
- T. Wu, Q. Yu, L. Hou, W. Duan, K. Wang and Q. Qin, J. Therm. Anal. Calorim., 2020, 140, 1837–1843 CrossRef CAS.
- T. Wu, Q. Yu, I. Roghair, K. Wang and M. van Sint Annaland, Chem. Eng. Process. - Process Intensif., 2020, 157, 108137 CrossRef CAS.
- A. Hameed, M. A. Gondal and Z. H. Yamani, Catal. Commun., 2004, 5, 715–719 CrossRef CAS.
- T. Otroshchenko, G. Jiang, V. A. Kondratenko, U. Rodemerck and E. V. Kondratenko, Chem. Soc. Rev., 2021, 50, 473–527 RSC.
- A. Khodakov, B. Olthof, A. T. Bell and E. Iglesia, J. Catal., 1999, 181, 205–216 CrossRef CAS.
- M. Puglisi, F. Arena, F. Frusteri, V. Sokolovskii and A. Parmaliana, Catal. Lett., 1996, 41, 41–43 CrossRef CAS.
- Y. Liu, Y. Cao, N. Yi, W. Feng, W. Dai, S. Yan, H. He and K. Fan, J. Catal., 2004, 224, 417–428 CrossRef CAS.
- Y. Liu, W. Feng, T. Li, H. He, W. Dai, W. Huang, Y. Cao and K. Fan, J. Catal., 2006, 239, 125–136 CrossRef CAS.
- Z. Han, X. Xue, J. Wu, W. Lang and Y. Guo, Chin. J. Catal., 2018, 39, 1099–1109 CrossRef CAS.
- M. J. Ndolomingo, N. Bingwa and R. Meijboom, J. Mater. Sci., 2020, 55, 6195–6241 CrossRef CAS.
- H. H. Kung, Advances in Catalysis, 1994, pp. 1–38 Search PubMed.
- J. T. Grant, C. A. Carrero, A. M. Love, R. Verel and I. Hermans, ACS Catal., 2015, 5, 5787–5793 CrossRef CAS.
- J. Schönherr, K. Eckl and H. Gruler, Planta, 1979, 147, 21–26 CrossRef PubMed.
- K. Chen, S. Xie, A. T. Bell and E. Iglesia, J. Catal., 2001, 198, 232–242 CrossRef CAS.
- A. Parmaliana, V. Sokolovskii, D. Miceli and N. Giordano, Appl. Catal., A, 1996, 135, L1–L5 CrossRef CAS.
- A. Corma, J. M. L. Nieto and N. Paredes, J. Catal., 1993, 144, 425–438 CrossRef CAS.
- X. T. Gao, P. Ruiz, Q. Xin, X. X. Guo and B. Delmon, J. Catal., 1994, 148, 56–67 CrossRef CAS.
- F. Ying, J. Li, C. Huang, W. Weng and H. Wan, Catal. Lett., 2007, 115, 137–142 CrossRef CAS.
- K. Chen, S. Xie, E. Iglesia and A. T. Bell, J. Catal., 2000, 189, 421–430 CrossRef CAS.
- K. Chen, S. Xie, A. T. Bell and E. Iglesia, J. Catal., 2000, 195, 244–252 CrossRef CAS.
- G. Lin, Y. Su, X. Duan and K. Xie, Angew. Chem., Int. Ed., 2021, 60, 9311–9315 CrossRef CAS PubMed.
- O. Lezla, E. Bordes, P. Courtine and G. Hecquet, J. Catal., 1997, 170, 346–356 CrossRef CAS.
- T. E. Davies, T. García, B. Solsona and S. H. Taylor, Chem. Commun., 2006, 3417–3419 RSC.
- S. Song, J. Li, Z. Wu, P. Zhang, Y. Sun, W. Song, Z. Li and J. Liu, AIChE J., 2022, 68, e17451 CrossRef CAS.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS PubMed.
- Z. Li, A. W. Peters, A. E. Platero-Prats, J. Liu, C.-W. Kung, H. Noh, M. R. DeStefano, N. M. Schweitzer, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2017, 139, 15251–15258 CrossRef CAS PubMed.
- M.-X. Huang, X. Wu, X.-D. Yi, G.-B. Han, W.-S. Xia and H.-L. Wan, RSC Adv., 2017, 7, 14846–14856 RSC.
- A. W. Peters, K. Otake, A. E. Platero-Prats, Z. Li, M. R. DeStefano, K. W. Chapman, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2018, 10, 15073–15078 CrossRef CAS PubMed.
- Y. Liu, R. Che, G. Chen, J. Fan, Z. Sun, Z. Wu, M. Wang, B. Li, J. Wei, Y. Wei, G. Wang, G. Guan, A. A. Elzatahry, A. A. Bagabas, A. M. Al-Enizi, Y. Deng, H. Peng and D. Zhao, Sci. Adv., 2015, 1, e1500166 CrossRef PubMed.
- J.-H. Li, C.-C. Wang, C.-J. Huang, Y.-F. Sun, W.-Z. Weng and H.-L. Wan, Appl. Catal., A, 2010, 382, 99–105 CrossRef CAS.
- Z. Teng, G. Zheng, Y. Dou, W. Li, C. Mou, X. Zhang, A. M. Asiri and D. Zhao, Angew. Chem., Int. Ed., 2012, 51, 2173–2177 CrossRef CAS PubMed.
- D. Li, H. Zhou and I. Honma, Nat. Mater., 2004, 3, 65–72 CrossRef CAS PubMed.
- X. Fan, D. Liu, Z. Zhao, J. Li and J. Liu, Catal. Today, 2020, 339, 67–78 CrossRef CAS.
- Y. Wu, Y. He, T. Chen, W. Weng and H. Wan, Appl. Surf. Sci., 2006, 252, 5220–5226 CrossRef CAS.
- J. Li, C. Wang, C. Huang, W. Weng and H. Wan, Catal. Lett., 2010, 137, 81–87 CrossRef CAS.
- Q. Xie, H. Zhang, J. Kang, J. Cheng, Q. Zhang and Y. Wang, ACS Catal., 2018, 8, 4902–4916 CrossRef CAS.
- Y. He, Y. Wu, T. Chen, W. Weng and H. Wan, Catal. Commun., 2006, 7, 268–271 CrossRef CAS.
- E. Heracleous and A. A. Lemonidou, J. Catal., 2010, 270, 67–75 CrossRef CAS.
- W. Cai-Cai, L. Jian-Hui, S. Yi-Fei, Z. Xiao-Quan, H. Chuan-Jing, W. Wei-Zheng and W. Hui-Lin, Acta Phys.-Chim. Sin., 2011, 27, 2421–2426 Search PubMed.
- A. P. Pushkar and J. J. Varghese, J. Catal., 2022, 413, 681–691 CrossRef CAS.
- B. Farin, P. Eloy, C. Poleunis, M. Devillers and E. M. Gaigneaux, Catal. Sci. Technol., 2016, 6, 6046–6056 RSC.
- L. Huai-Qian, S. Lei, H. Chong, W. Wei-Zheng, H. Chuan-Jing and W. Hui-Lin, Acta Phys.-Chim. Sin., 2012, 28, 2697–2704 Search PubMed.
- P. Boizumault-Moriceau, Appl. Catal., A, 2003, 245, 55–67 CrossRef CAS.
- H. Alasiri, S. Ahmed, F. Rahman, A. Al-Amer and U. B. Majeed, Can. J. Chem. Eng., 2019, 97, 2340–2346 CrossRef CAS.
- K. Shimoda, S. Ishikawa, K. Matsumoto, M. Miyasawa, M. Takebe, R. Matsumoto, S. Lee and W. Ueda, ChemCatChem, 2021, 13, 3132–3139 CrossRef CAS.
- A. Massó Ramírez, F. Ivars-Barceló and J. M. López Nieto, Catal. Today, 2020, 356, 322–329 CrossRef.
- L. Schumacher, J. Pfeiffer, J. Shen, T. Gutmann, H. Breitzke, G. Buntkowsky, K. Hofmann and C. Hess, ACS Catal., 2023, 13, 8139–8160 CrossRef CAS.
- A. Bielaski, J. Catal., 1972, 25, 398–406 CrossRef.
- X. Zhang, J. Liu, Y. Jing and Y. Xie, Appl. Catal., A, 2003, 240, 143–150 CrossRef CAS.
- Y. Schuurman, V. Ducarme, T. Chen, W. Li, C. Mirodatos and G. A. Martin, Appl. Catal., A, 1997, 163, 227–235 CrossRef CAS.
- Q. Zhang, C. Cao, T. Xu, M. Sun, J. Zhang, Y. Wang and H. Wan, Chem. Commun., 2009, 2376 RSC.
- E. Heracleous and A. Lemonidou, J. Catal., 2006, 237, 162–174 CrossRef CAS.
- E. Cartuyvels, G. Absillis and T. N. Parac-Vogt, Chem. Commun., 2008, 85–87 RSC.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837 RSC.
- A. Beretta, L. Piovesan and P. Forzatti, J. Catal., 1999, 184, 455–468 CrossRef CAS.
- X. Gao, L. Zhu, F. Yang, L. Zhang, W. Xu, X. Zhou, Y. Huang, H. Song, L. Lin, X. Wen, D. Ma and S. Yao, Nat. Commun., 2023, 14, 1478 CrossRef CAS PubMed.
- M. B. Gawande, K. Ariga and Y. Yamauchi, Small, 2021, 17, 2101584 CrossRef CAS PubMed.
- J. Li, M. F. Stephanopoulos and Y. Xia, Chem. Rev., 2020, 120, 11699–11702 CrossRef CAS PubMed.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- Y. Xing, L. Kang, J. Ma, Q. Jiang, Y. Su, S. Zhang, X. Xu, L. Li, A. Wang, Z.-P. Liu, S. Ma, X. Y. Liu and T. Zhang, Chin. J. Catal., 2023, 48, 164–174 CrossRef CAS.
- Q. Zhang, X. Jiang, Y. Li, Y. Tan, Q. Jiang, X. Liu and B. Qiao, Chin. J. Chem., 2024, 42, 370–376 CrossRef CAS.
- R. Peng, S. Li, X. Sun, Q. Ren, L. Chen, M. Fu, J. Wu and D. Ye, Appl. Catal., B, 2018, 220, 462–470 CrossRef CAS.
- Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo and G. Lu, ACS Catal., 2016, 6, 2265–2279 CrossRef CAS.
- J. Liu, M. Hao, C. Chen, K. Du, Q. Zhou, S. Zou, L. Xiao and J. Fan, Appl. Surf. Sci., 2020, 528, 147025 CrossRef CAS.
- H. L. Wan, X. P. Zhou, W. Z. Weng, R. Q. Long, Z. S. Chao, W. De Zhang, M. S. Chen, J. Z. Luo and S. Q. Zhou, Catal. Today, 1999, 51, 161–175 CrossRef CAS.
- I. Trotuş, C. M. Teodorescu, V. I. Pârvulescu and I. Marcu, ChemCatChem, 2013, 5, 757–765 CrossRef.
- J. M. Venegas, W. P. McDermott and I. Hermans, Acc. Chem. Res., 2018, 51, 2556–2564 CrossRef CAS PubMed.
- C. Xu, C. Ge, D. Sun, Y. Fan and X.-B. Wang, Nanotechnology, 2022, 33, 432003 CrossRef CAS PubMed.
- X. Jiang, K. Zhang, M. J. Forte, S. Cao, B. S. Hanna and Z. Wu, Catal. Rev., 2022, 1–80 Search PubMed.
- H. Chen, D. Jiang, Z. Yang and S. Dai, Acc. Chem. Res., 2023, 56, 52–65 CrossRef CAS PubMed.
- Z. Fu, D.-Z. Li, L.-D. Zhou, Y.-M. Li, J.-W. Guo, Y.-Q. Li, H.-M. Liu and Q.-J. Zhang, Pet. Sci., 2023, 20, 2488–2498 CrossRef CAS.
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Science, 2016, 354, 1570–1573 CrossRef CAS PubMed.
- L. Shi, D. Wang, W. Song, D. Shao, W. Zhang and A. Lu, ChemCatChem, 2017, 9, 1788–1793 CrossRef CAS.
- P. Chaturbedy, M. Ahamed and M. Eswaramoorthy, ACS Omega, 2018, 3, 369–374 CrossRef CAS PubMed.
- G. Wang, X. Zhang, Y. Yan, X. Huang and Z. Xie, Appl. Catal., A, 2021, 628, 118402 CrossRef CAS.
- Z. Liu, B. Yan, S. Meng, R. Liu, W. Lu, J. Sheng, Y. Yi and A. Lu, Angew. Chem., Int. Ed., 2021, 60, 19691–19695 CrossRef CAS PubMed.
- A. M. Love, B. Thomas, S. E. Specht, M. P. Hanrahan, J. M. Venegas, S. P. Burt, J. T. Grant, M. C. Cendejas, W. P. McDermott, A. J. Rossini and I. Hermans, J. Am. Chem. Soc., 2019, 141, 182–190 CrossRef CAS PubMed.
- G. Wang, Y. Yan, X. Zhang, X. Gao and Z. Xie, Ind. Eng. Chem. Res., 2021, 60, 17949–17958 CrossRef CAS.
- J. T. Grant, W. P. McDermott, J. M. Venegas, S. P. Burt, J. Micka, S. P. Phivilay, C. A. Carrero and I. Hermans, ChemCatChem, 2017, 9, 3623–3626 CrossRef CAS.
- L. Shi, D. Wang and A. Lu, Chin. J. Catal., 2018, 39, 908–913 CrossRef CAS.
- L. Shi, Y. Wang, B. Yan, W. Song, D. Shao and A.-H. Lu, Chem. Commun., 2018, 54, 10936–10946 RSC.
- D. Creaser, B. Andersson, R. R. Hudgins and P. L. Silveston, Chem. Eng. Sci., 1999, 54, 4365–4370 CrossRef CAS.
- H. Li, J. Zhang, P. Wu, S. Xun, W. Jiang, M. Zhang, W. Zhu and H. Li, J. Phys. Chem. C, 2019, 123, 2256–2266 CrossRef CAS.
- B. Rajbanshi, S. Saha, C. Fricke, S. C. Ammal and A. Heyden, Catal. Sci. Technol., 2020, 10, 5181–5195 RSC.
- G. Wang, S. Chen, Q. Duan, F. Wei, S. Lin and Z. Xie, Angew. Chem., Int. Ed., 2023, 62, e202307470 CrossRef CAS PubMed.
- Q. Zhou, Z. Liu, L. Zhu, W. Lu, L. He and D. Wang, J. Phys. Chem. C, 2023, 127, 12942–12952 CrossRef CAS.
- L. Zhu, Z. Liu, Q. Zhou, W.-D. Lu and D. Wang, J. Phys. Chem. C, 2023, 127, 6280–6293 CrossRef CAS.
- F. Wu, Z. Liu, J. Sheng, L. Zhu, W.-D. Lu, B. Qiu, D. Wang and A.-H. Lu, J. Catal., 2023, 424, 121–129 CrossRef CAS.
- B. Frank, J. Zhang, R. Blume, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2009, 48, 6913–6917 CrossRef CAS PubMed.
- Z. Sui, J. Zhou, Y. Dai and W. Yuan, Catal. Today, 2005, 106, 90–94 CrossRef CAS.
- X. Sun, Y. Ding, B. Zhang, R. Huang and D. S. Su, Chem. Commun., 2015, 51, 9145–9148 RSC.
- C. Chen, J. Zhang, B. Zhang, C. Yu, F. Peng and D. Su, Chem. Commun., 2013, 49, 8151 RSC.
- T. Cao, X. Dai, W. Liu, Y. Fu and W. Qi, Carbon N. Y., 2022, 189, 199–209 CrossRef CAS.
- L. Cao, P. Dai, L. Zhu, L. Yan, R. Chen, D. Liu, X. Gu, L. Li, Q. Xue and X. Zhao, Appl. Catal., B, 2020, 262, 118277 CrossRef.
- D. Yang, D. Liu, Y. Li, H. Gan, P. Xu, Y. Tian, Z. Li, T. Xing, X. Gu, L. Li, X. Wang, L. Wei, P. Dai and M. Wu, Appl. Surf. Sci., 2023, 639, 158258 CrossRef CAS.
- X. Sun, P. Han, B. Li, S. Mao, T. Liu, S. Ali, Z. Lian and D. Su, Chem. Commun., 2018, 54, 864–875 RSC.
- W. Qi and D. Su, ACS Catal., 2014, 4, 3212–3218 CrossRef CAS.
- C. Li, H. Zhang, W. Liu, L. Sheng, M.-J. Cheng, B. Xu, G. Luo and Q. Lu, Nat. Commun., 2024, 15, 884 CrossRef CAS PubMed.
- K. Zhang, S. Sun and K. Huang, Chem. Eng. J., 2024, 481, 148395 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |