
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in transition metal complexes featuring silylene as ligands
Zohreh
Hendi†
,
Madhusudan K.
Pandey†
 ,
Saroj Kumar
Kushvaha
and
Herbert W.
Roesky
*
,
Saroj Kumar
Kushvaha
and
Herbert W.
Roesky
*
Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Göttingen, 37077, Germany. E-mail: hroesky@gwdg.de
First published on 29th June 2024
Abstract
Silylenes, divalent silicon(II) compounds, once considered highly reactive and transient species, are now widely employed as stable synthons in main-group and coordination chemistry for myriad applications. The synthesis of stable silylenes represents a major breakthrough, which led to extensive exploration of silylenes in stabilizing low-valent main-group elements and as versatile ligands in coordination chemistry and catalysis. In recent years, the exploration of transition metal complexes stabilized with silylene ligands has captivated significant research attention. This is due to their robust σ-donor characteristics and capacity to stabilize transition metals in low valent states. It has also been demonstrated that the transition metal complexes of silylenes are effective catalysts for hydroboration, hydrosilylation, hydrogenation, hydrogen isotope exchange reactions, and small molecule activation chemistry. This review article focuses on the recent progress in the synthesis and catalytic application of transition metal complexes of silylenes.
Zohreh Hendi received her MSc degree from the Iran University of Science and Technology and her PhD degree from the Sharif University of Technology, Tehran, Iran. in 2022, where she worked with Prof. Sirous Jamali studying synthesis and photophysical properties of naphthalimide-based NHC ligands and its coinage metal complexes in addition to platinum-based metal–organic frameworks. After completing her PhD, she moved to Germany for her post-doctoral work with Prof. Herbert W. Roesky at Georg-August-Universität Göttingen, Germany. Her current research includes the synthesis of compounds with low-valent silicon and their exploration in small molecule activation reactions. |
Madhusudan Kumar Pandey obtained his BSc and MSc degrees from B. R. A. B. U. Muzaffarpur and Pondicherry University. He received his doctoral degree in 2019 from IIT Bombay under the supervision of Prof. M. S. Balakrishna, studying sterically demanding phosphines and their transition metal complexes for catalytic applications. He was a postdoctoral research associate in the group of Prof. Joyanta Choudhury at IISER Bhopal, India (2020–2022). Currently, he is working as a postdoctoral researcher with Prof. Herbert W. Roesky at Georg-August-Universität Göttingen, Germany. His current research interest includes synthesizing compounds with low-valent silicon to stabilize main group elements in unusual oxidation states. |
Saroj Kumar Kushvaha completed his BSc from the University of Lucknow in 2010 and MSc from DAVV Indore in 2012. Subsequently, he worked as a chemistry lecturer from 2013–2016. He worked as a junior researcher at Defence research and development organization (DRDO) for a short period of time. He received his PhD from Indian Institute of Technology Madras, Chennai, in 2022. Currently, he is working as a post-doctoral researcher at Georg-August-Universität Göttingen, Germany, He works in the area of main-group chemistry and molecular magnetism. |
Herbert W. Roesky earned his doctorate from the University of Göttingen. Following a stint at Du Pont in the United States, he returned to Göttingen to complete his habilitation. By 1971, he had assumed a professorship at Johann-Wolfgang-Goethe-Universität in Frankfurt am Main. Later, in 1980, he transitioned to the University of Göttingen, where he served as the director of the Institute for Inorganic Chemistry until 2004. Roesky is renowned for his ground-breaking research on fluorides across transition and main group elements. Presently, his focus lies on various facets of compounds featuring low-valent silicon. His prolific contributions include over 1350 peer-reviewed papers, articles, patents, and books spanning the realms of Inorganic Chemistry and Material Sciences. |
Introduction
Developing task-specific ligands is vital in various fields of chemistry, such as organometallic chemistry, catalysis, and material chemistry. The need for novel ligands has always been a key area of research because chemists are always curious to learn about new compounds and ways to improve the properties and processes of already existing ones.1 The enormous significance of phosphine ligands in various fields of chemistry also suggests that one can tailor-make task-specific ligands for various applications.2 Over the past few years, fundamental and application-oriented transition-metal chemistry has seen a surge in studying the ligand characteristics of stable N-heterocyclic carbenes (NHCs) over phosphines. NHCs are considered excellent ligands due to their strong σ-donor and weak π-acceptor properties, facilitating strong bonding interactions with metals and main-group elements. They are widely utilized in main-group chemistry, coordination chemistry, and catalysis.3 The fast growth of NHC-based transition-metal complexes has influenced scientists to discover and explore the ligating capabilities of heavier carbene analogues, such as silylene.Silylenes, heavier analogues of carbenes, feature a Si(II) atom with one lone pair of electrons and a vacant 3p orbital, rendering them potential σ-donor and π-acceptor ligands for transition metals. In 1937, Schwarz and Pietsch reported the first divalent Si(II) compound, Cl2Si:, by reducing SiCl4 through glow discharge.4 After this, dimethylsilylene (Me2Si:) was observed at low-temperature in argon matrices and was considered a reactive intermediate in the chemical reactions.5 Silylenes (R2Si:), were often regarded as very reactive intermediates that lost their identity by various mechanisms, including cycloaddition, polymerization, insertion, and oligomerization, which prevented silylenes from being utilized as reactants in laboratory experiments.6 It was observed that silylenes with a small R substituent are unstable.5 In contrast, disilene formation was observed when the steric bulk of the R substituent was increased.7 This suggests that kinetic and/or thermodynamic stabilization is essential to isolate silylene (R2Si:) as a stable compound. In 1986, Jutzi et al. made a ground-breaking discovery in silylene chemistry by isolating decamethyl silicocene (η5-C5Me5)2Si: (I), the first stable compound featuring divalent silicon(II) atom (Fig. 1).8 This discovery demonstrated that electronic and steric saturation is necessary for synthesizing bottleable silylenes. The first significant breakthrough in silylene chemistry occurred with synthesis of the first N-heterocyclic silylene (NHSi) II in 1994 by West and Denk (Fig. 1).9 Following this, divalent silicon compound chemistry saw a rapid expansion. Since the mid-1990s, numerous stable cyclic silylenes III–V with diverse substitution patterns and ring sizes have been reported.6,10 However, all these stable silylenes contain divalent Si(II) atoms with no further scope for functionalization. In 2006, Roesky and co-workers isolated a unique heteroleptic three-coordinate chlorosilylene VI.11 Following this in 2009, Roesky and Filipou groups independently synthesized NHC-stabilized X2Si: VII12 silylenes with functionalizable Si–X (X = Cl, Br) bonds (Fig. 1).
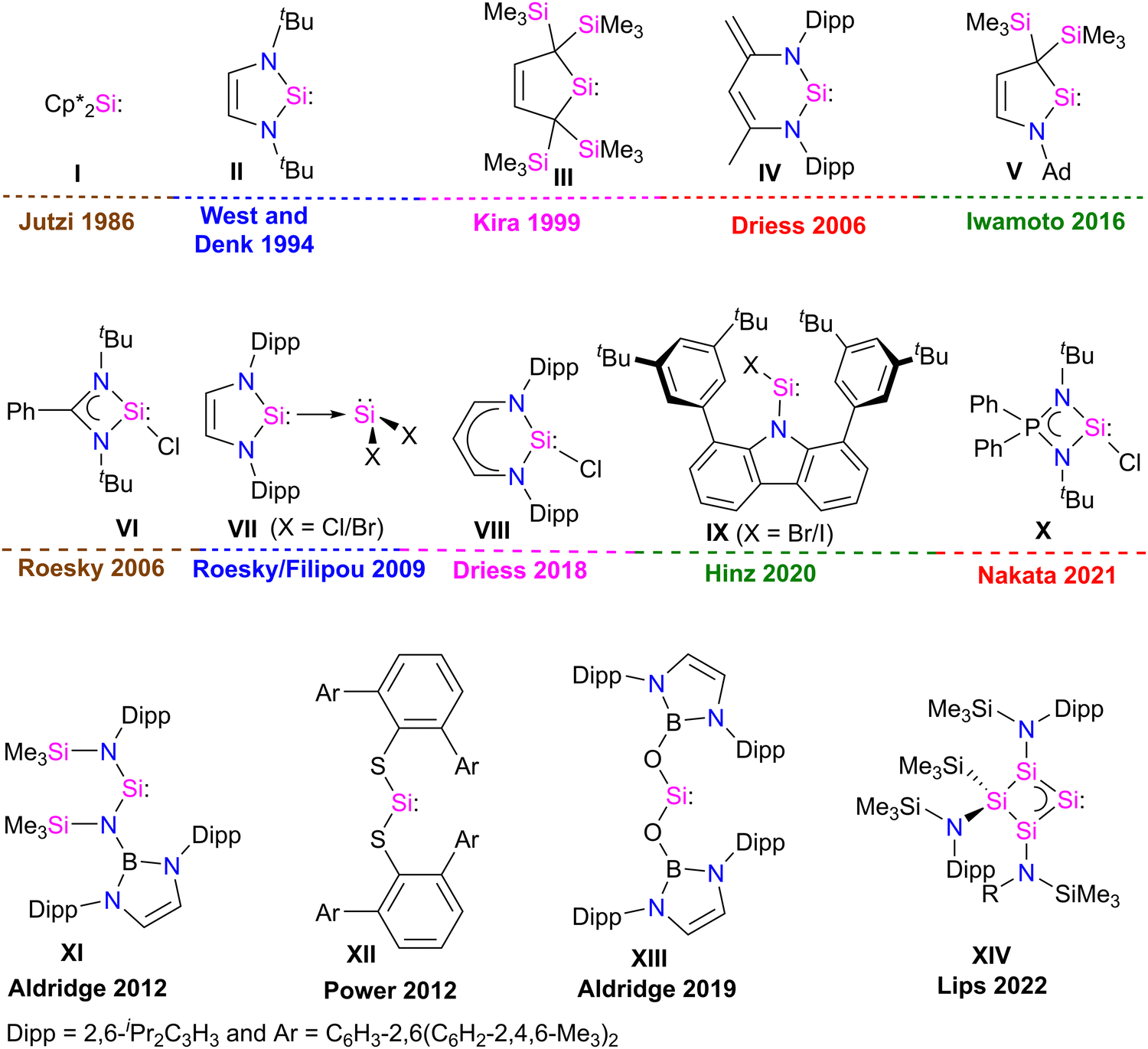 | ||
| Fig. 1 Important breakthrough in silylene chemistry over the years. | ||
A three-coordinate chlorosilylene VIII stabilized by β-diketiminate ligand was also isolated by Driess in 2018.13 Very recently, Hinz isolated a unique two-coordinate chlorosilylene IX stabilized by a bulky carbazole ligand.14 Nakata isolated a strong σ-donor three coordinate iminophosphonamido–chlorosilylene X in 2021.15 These compounds have been widely employed in silylene chemistry for many years after these discoveries.16,17 Recently, the Power, Jones, Aldridge, and Inoue groups developed a unique class of two coordinate acyclic silylenes XI–XIII, and Lips and co-workers developed a homocyclic silylene XIV with small HOMO–LUMO gap.18 Two-coordinate acyclic silylenes, owing to vacant 3p orbital at the Si atom and small HOMO–LUMO gap, are very reactive and act as transition metal mimics in small molecule activation chemistry.18 These discoveries changed the course of silylene chemistry and started a burgeoning era.
Transition metal complexes of silylenes: a historical perspective
Unlike NHC, transition metal complexes of NHSis are still in the early stages of development. The exploration of transition metal complexes of NHSis lags behind the extensive utilization of NHC-stabilized transition metal complexes in various important applications.19 The lack of progress is mostly due to the restricted techniques available for synthesizing silylene transition metal complexes and their instability. The syntheses of silylene complexes via a 1,2-hydride shift from a silyl ligand to the transition metals are known.19e,f However, unlike the trans-metalation and the base-mediated procedures to synthesize NHC-stabilized transition metal complexes, the primary method for synthesizing silylene transition metal complexes involve free silylene coordination, which restricts their production and use. In 1977, Welz and Schmid unveiled the first example of a thermolabile Fe complex of silylene, which was only stable below −20 °C.20 However, in 1987, a breakthrough came when Zybill and Müller achieved the first structural characterization of the silylene transition metal complex, promising further development.21 Their methodology involved treating Fe(II) precursors [K2(Fe(CO)4)] or [H2(Fe(CO)4)] with (tBuO)2SiCl2 under salt metathesis conditions or with a base (Et3N), to produce the first pioneering silylene Fe complex.The landscape of this field transformed significantly with the synthesis of the first bottleable NHSis in 1994, following which several reports emerged detailing silylene complexes with various transition metals. These advancements and early breakthroughs were succinctly compiled and reviewed in a comprehensive report by West and co-workers.6 The synthesis of three-coordinate chlorosilylene by Roesky and co-workers significantly boosted the development of transition metal complexes of base-stabilized silylenes. The presence of the Si–Cl bond enabled the synthesis of various functionalized silylenes, leading to the exploration of their transition metal chemistry in diverse avenues.16a–c As a result, the past few years have seen a surge of interest in the synthesis of transition metal complexes of various silylene ligands and their utility in various catalytic applications (Fig. 2).17c,22 The presence of silylene in this class of compounds, as potent σ-donor ligands, fine-tune the catalytic activity of the metal center in metal-mediated homogeneous catalysis.
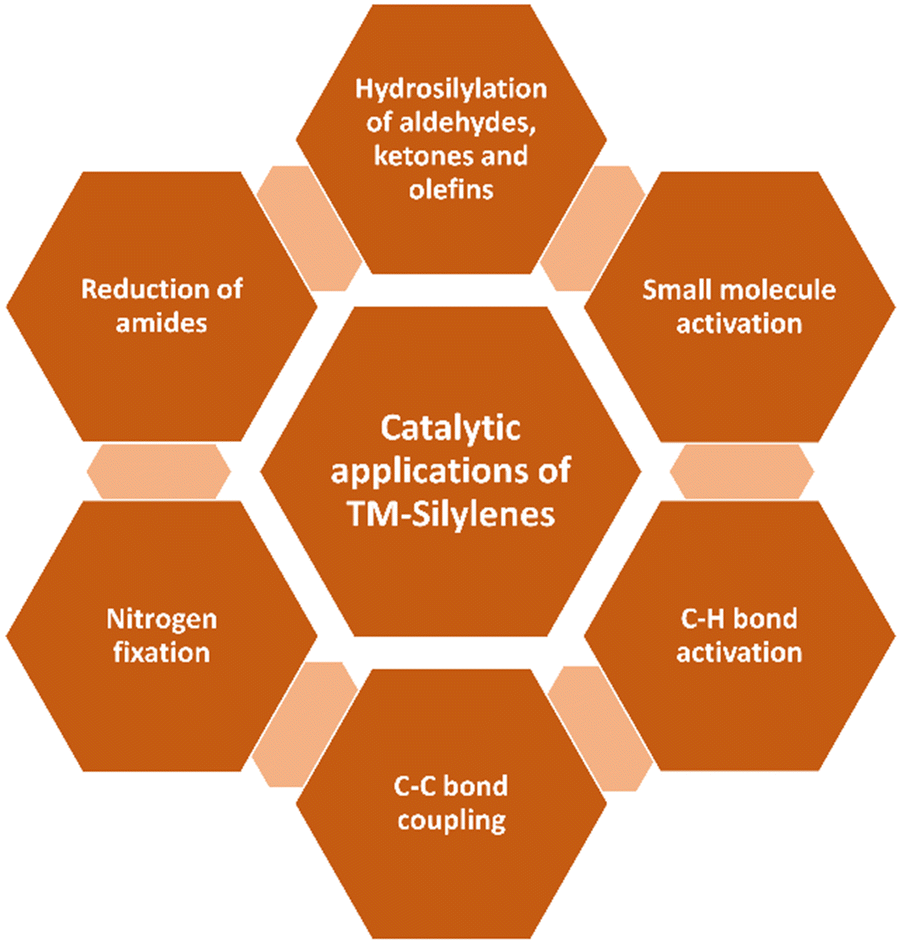 | ||
| Fig. 2 Catalytic applications of transition metal silylene complexes. | ||
Driess and co-workers were the first to summarize the utility of transition metal complexes of silylenes in various catalytic reactions.17d,22a Recently, Li and co-workers summarized the progress in the synthesis and characterization of Fe, Co, and Ni complexes of silylenes.17b In 2021, Khan and co-workers surveyed the coinage metal complexes of silylenes.17a Although these elegant reviews provide a state-of-the-art description of silylene-transition metal complexes, there is a need to survey the recent advancements made in the field from 2021. This review aims to bridge the gap by summarizing recent advancements in the synthesis, characterization, and catalytic utilities of transition metal complexes of silylenes reported after 2021. Through a systematic literature analysis, this review highlights key findings, discusses synthetic strategies, and evaluates catalytic performances, providing valuable insights for future research in this rapidly evolving field.
Fe, Co, and Mn complexes of silylenes
Unlike other transition metals, the report on Fe silylene complexes is mature.17d Some of the early developments in the field included the first spectroscopic characterization of Fe silylene complex XV in 1977.20 Followed by the first structural characterization of Fe silylene complex XVI in 1987.21 Subsequently, West reported the silylene coordinated Fe(0) complex XVII in 2001.23 In 2009, Roesky and co-workers introduced the first stable Fe complex XVIII, featuring a base-stabilized tricoordinate silylene ligand.24 Following which, several Fe–silylene complexes are synthesized and characterized (XIX–XXV: Fig. 3).17b,d The Fe complexes of silylenes are active in important catalytic reactions such as hydroboration and hydrosilylation of carbonyl compounds25 and the reductive functionalization of dinitrogen.26 Unlike Fe, the report on the Co and Mn complexes of silylenes is not extensive. However, recent years have seen a surge in the synthesis of Co and Mn complexes stabilized by silylenes, which are found to be important catalysts for the hydroboration of aldehyde and ketones and in the reductive functionalization of highly inert dinitrogen gas.17b Some of the earlier reported Co and Mn complexes are shown in Fig. 3 (XXVI–XXXII).17b,d,27 In this section, we will discuss the synthesis and application of Fe, Co, and Mn silylene complexes, focusing on recent advances from 2021 to the present date.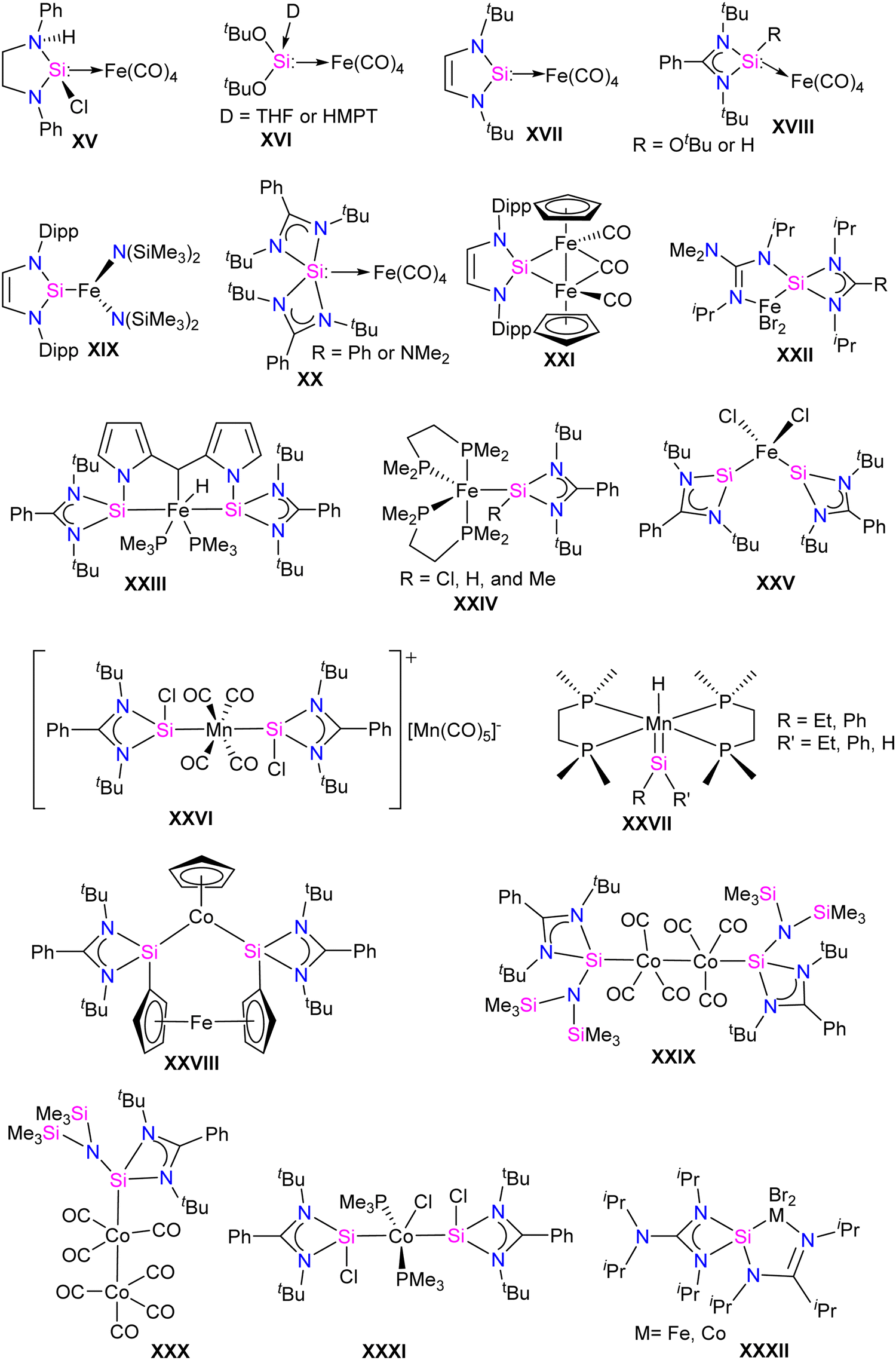 | ||
| Fig. 3 Selected examples of Fe, Co, and Mn–silylene complexes. | ||
Fe–silylene complexes
Recently, there has been significant progress in CO activation chemistry, including low-valent elements from Group 13 and Group 14.28 Among many classes of main group compounds that exhibit the ability to activate CO, silicon(I) and silicon(II) compounds have been extensively researched because of their small HOMO–LUMO energy gap, the presence of a lone pair, and a free p-orbital at the silicon center. The amidinate stabilized three coordinate bis-silylenes [{PhC(NtBu)2}Si]2 developed by Roesky and co-workers represent one of the most sought-after systems in silylene chemistry.16a The presence of a lone pair of electrons at each Si(I) atom and a dynamic Si(I)–Si(I) bond make this compound highly reactive; as a result, a remarkable array of reactions is known with this system.16b Nevertheless, the activation of CO has not been investigated using this class of compound until recently.Given this, Jones and co-workers explored the reactivity of an interconnected bis(silylene) 1 in the presence of CO gas. The reaction of 1 with CO gas (1 atm.) led to the cleavage of the Si–Si bond and the subsequent insertion of CO into the N–Si bond of one of the four-membered CNSi2 ring of bis(silylene) 1, leading to the formation of a novel bis(silylene) compound 2 (Scheme 1).29 The product, 2, can be formulated as a spacer-separated bis(silylene) and comprises an O atom bridge that connects one four-membered silylene fragment with a five-membered silylene ring. The unique five-membered silylene ring is the first example of a silicon analog of an “abnormal” N-heterocyclic carbene (aNHC). The 29Si NMR spectrum of 2 showed two singlet resonances at 35.2 and −12.2 ppm that are significantly upfield shifted compared to 1 (δ = 96.9 ppm).
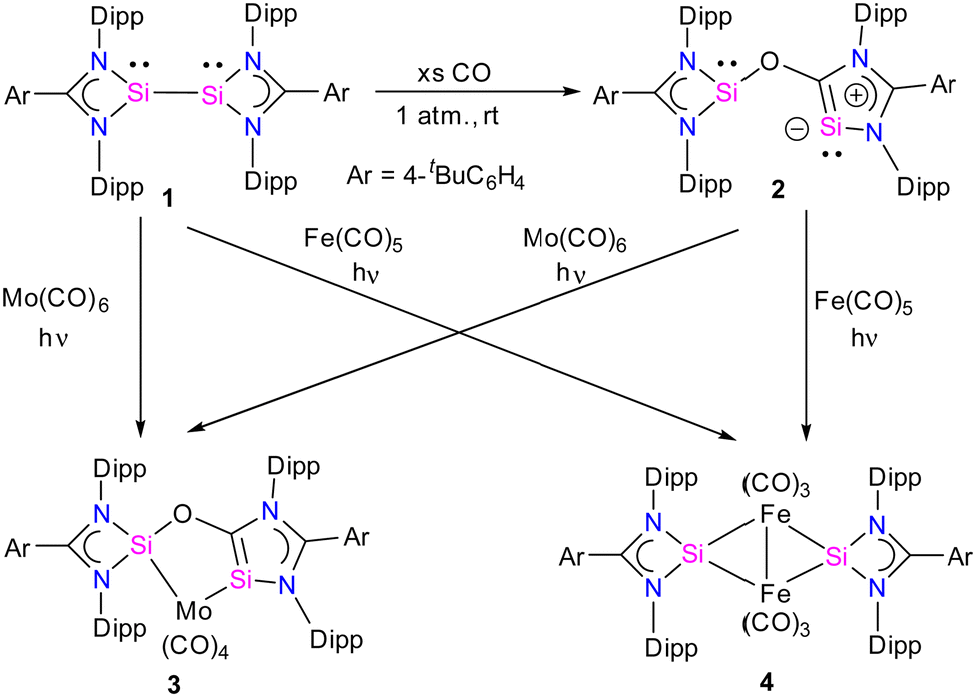 | ||
| Scheme 1 Synthesis of 2–4. | ||
The molecular structure of compound 2 revealed a distance of 3.441(1) Å between two- and three-coordinate silicon atoms, thus ruling out any significant bonding interaction (Fig. 4). As a result, compound 2 has the potential to function as a bidentate ligand in coordination chemistry. The bis-silylene 2 did not undergo a reaction with Mo(CO)6 or Fe(CO)5 at room temperature. However, when a solution of 2 in toluene or benzene, together with Mo(CO)6 or Fe(CO)5, was exposed to UV light from an LED lamp (370 nm, 43 W) for two hours, resulted in the formation of a mono-nuclear κ2-Si molybdenum complex 3 and a silyleneyl-bridged iron complex 4, respectively. Interestingly, compound 3 was also obtained by irradiating a mixture of 1 and Mo(CO)6 with a UV lamp for two hours. This indicates that 1 initially reacts with CO released from Mo(CO)6, forming 2. Subsequently, compound 2 reacts with Mo(CO)6 to produce the κ2-Si molybdenum complex 3. In the case of complex 4, the reaction proceeds with the release of CO molecules from 2 under UV irradiation to afford 1, which further reacts with Fe(CO)5 to afford silyleneyl-bridged iron complex 4. The control experiments further demonstrated this, which showed that a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1 and 2 resulted from irradiating a solution of 2. Furthermore, compound 4 was formed when 1 reacted with two equivalents of Fe(CO)5 under UV light. These control experiments suggest that the CO release from 2 is the first step during the reaction of 2 with Fe(CO)5.
:1 mixture of 1 and 2 resulted from irradiating a solution of 2. Furthermore, compound 4 was formed when 1 reacted with two equivalents of Fe(CO)5 under UV light. These control experiments suggest that the CO release from 2 is the first step during the reaction of 2 with Fe(CO)5.
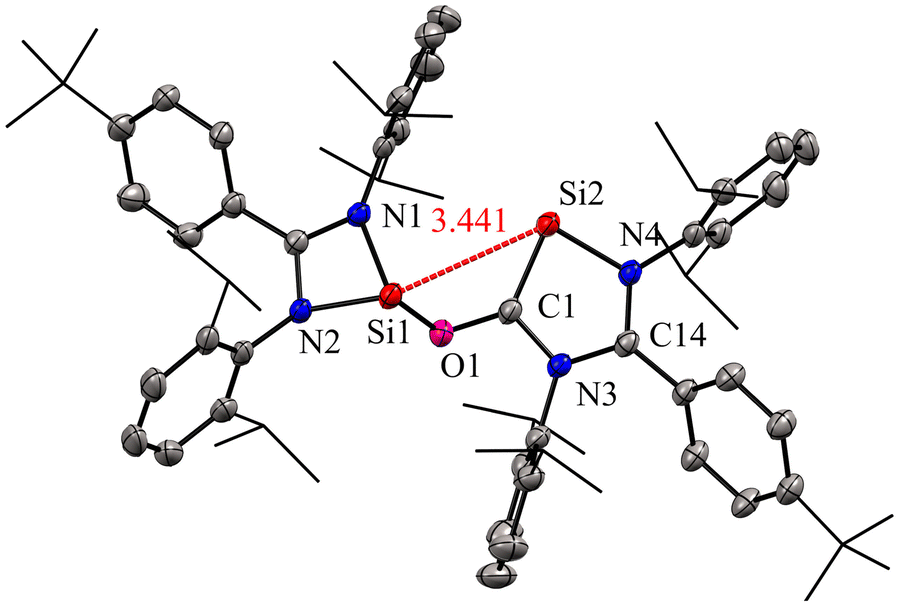 | ||
| Fig. 4 Molecular structure of 2 showing Si⋯Si distance between two coordinate and three coordinate Si atoms. Reproduced from ref. 29 ©2022 Wiley-VCH GmbH. | ||
Further, the molecular structure of 4 also revealed, formally three-electron donor, silyleneyl ligands are bridging the (CO)3Fe–Fe(CO)3 moiety, yielding an 18-electron complex. The IR spectrum of complex 3 showed CO stretching vibrations at (ν = 2005, 1900, 1870, 1835 cm−1), which are higher than the same reported for chelating bis(NHC)Mo(CO)4 complexes,30 hence suggesting the bis-silylene 2 to be less σ-donor than NHCs or aNHCs. The authors have also done detailed DFT calculations to understand the formation of 2 from 1. The reaction starts with the initial coordination of CO to one Si atom of 1 to yield Int1. Next, viaTS1 (11.2 kcal mol−1), the coordinated CO inserts into a Si–N bond to produce Int2. Attack of the O-atom of Int2 to the four-membered silylene Si atom occurs viaTS2 (4.7 kcal mol−1), leading to the cleavage of the Si–Si bond to give 2 (Fig. 5).
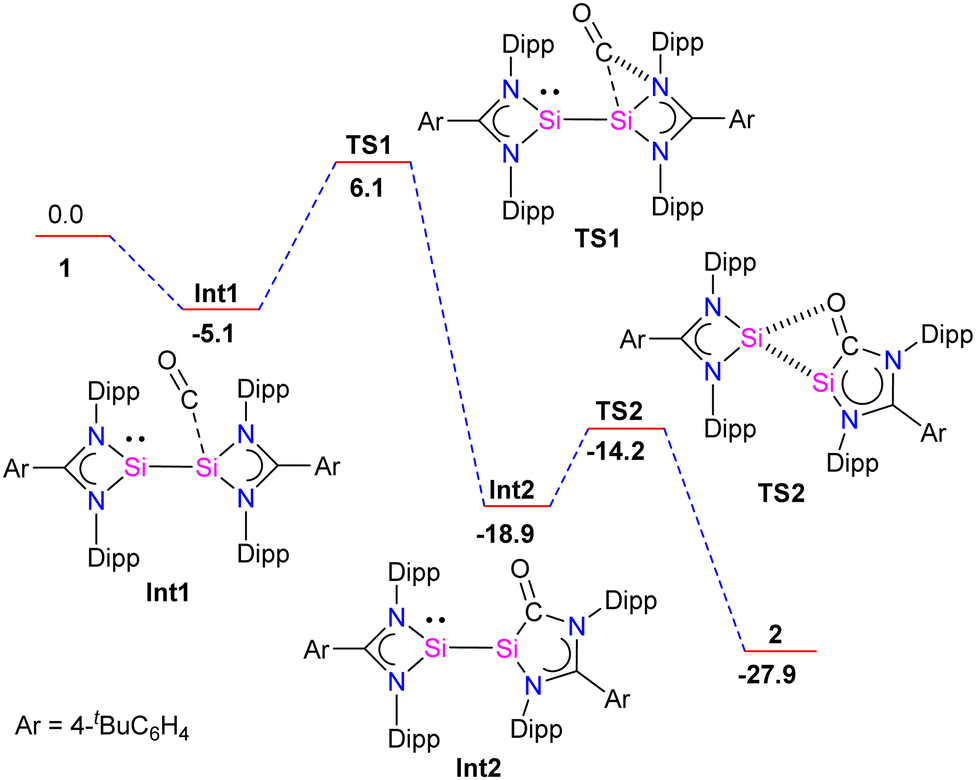 | ||
| Fig. 5 DFT calculated probable mechanism for the formation of 2 (energies are expressed as kcal mol−1, Ar = 4-tBuC6H4). The calculation was done with Gaussian09 using the hybrid functional B3PW91.6-31G(d,p), double-ζ basis set was used for C, H, O and N atoms, whereas Si atoms were represented with a small-core StuttgartDresden relativistic effective core potential associated with their adapted basis set. | ||
Bis(silylene)s are categorized into two types: (i) interconnected bis(silylene)s, where two Si(I) atoms are directly connected. (ii) spacer-separated bis(silylene)s, wherein a spacer motive connects two Si(II) centers.16b Reactivity studies of interconnected bis(silylene)s with transition metals, aimed at forming mononuclear metal complexes, typically result in Si–Si bond cleavage (Scheme 2(a)). This phenomenon is also exemplified in the reaction of 1 with Fe(CO)5, separating two silicon centers and forming 4. However, it is worth noting that there are instances where the formation of multinuclear transition metal complexes with interconnected bis(silylene)s has been observed.31 Krogman and co-workers reported the first mononuclear metal complex 7 of interconnected bis(silylene) by altering the reaction pathway, wherein the Si–Si bond can induce η2-coordination to the metal center (Scheme 2).32 A two-step reduction of the chlorosilylene-stabilized Fe(II) complex 5 with KC8 produced the mononuclear metallacyclic complex 7, characterized by a direct Si–Si bond. The molecular structure of 7 (Fig. 6) shows that Si(I)–Fe–Si(I) atoms form a three-membered equilateral triangle with Si–Si bond distance of 2.217(6) Å, which falls in the range of Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si double bonds (2.120–2.250 Å).33 Complex 7 exhibits an unconventional electronic structure, where the three-membered Fe–Si–Si ring displays 2π-aromaticity. DFT calculations on this compound unveiled an electronic configuration where the Si–Si fragment acts as a four-electron σ-donor to the Fe center.
Si double bonds (2.120–2.250 Å).33 Complex 7 exhibits an unconventional electronic structure, where the three-membered Fe–Si–Si ring displays 2π-aromaticity. DFT calculations on this compound unveiled an electronic configuration where the Si–Si fragment acts as a four-electron σ-donor to the Fe center.
 | ||
| Scheme 2 (a) Non-reactivity of interconnected bis(silylene) for the formation of mononuclear metal adduct to Si–Si bond (b) Synthetic of complex 7. | ||
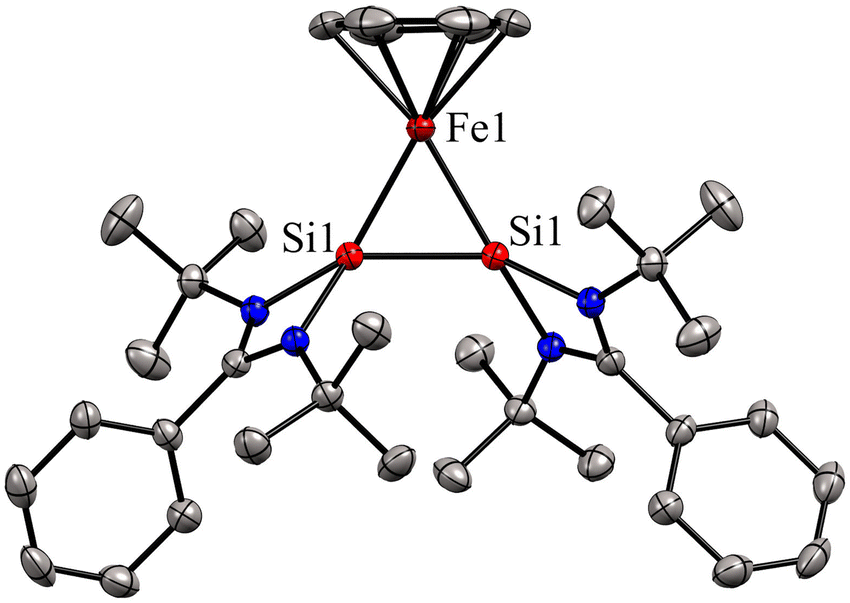 | ||
| Fig. 6 Molecular structure of 7. Reproduced from ref. 32 Copyright©2022, American Chemical Society. | ||
Furthermore, a considerable π-back donation from the Fe(0) center to the silicon atoms within the disilylene moiety enhances the overall structural stabilization. The reactivity of compound 7 was assessed through its reaction with trimethylsilylazide and benzophenone (Scheme 3). The reaction of compound 7 with trimethylsilylazide at 40 °C resulted in the insertion of azide into the Si–Si bond. According to DFT calculations, the mechanism of this reaction can be elucidated by the capability of 7 to exhibit nucleophilic-induced FLP (Frustrated Lewis Pair) reactivity. This means that the Si(I) centers in complex 7 have the dual ability to act as nucleophiles and electrophiles. In the first step, the azide undergoes a nucleophilic attack on one of the Si centers, inducing a nucleophilic character at the second Si center. Subsequently, this nucleophilic Si center attacks back onto the azide, forming 8 (Fig. 7).
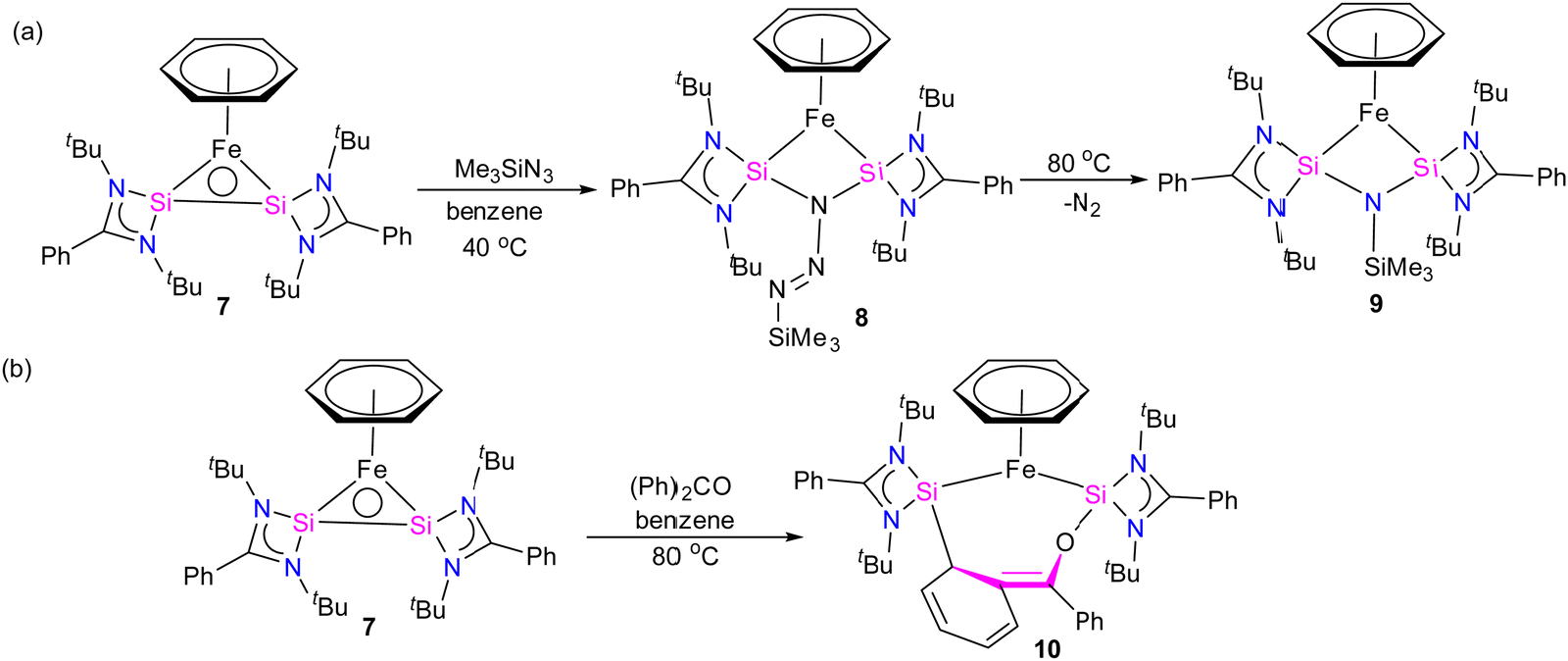 | ||
| Scheme 3 The reaction of 7 with (a) Me3SiN3 and (b) benzophenone. | ||
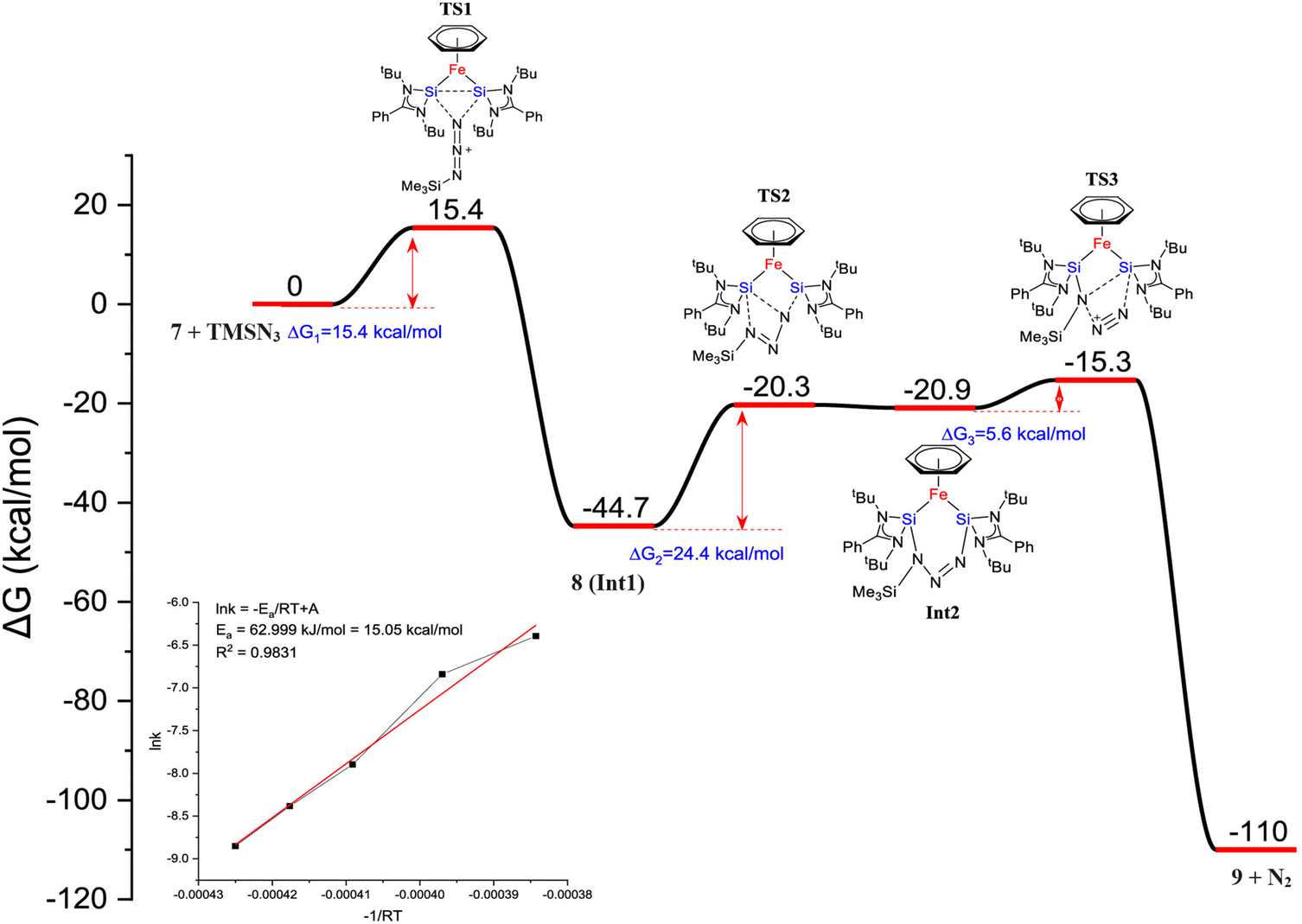 | ||
| Fig. 7 DFT calculated free energy profile for the formation of 9 from trimethylsilyl azide and complex 7 (B3LYP-TZVP/def2-TZVP). The consumption of 7 is shown in the inset. (Reproduced from ref. 32 Copyright©2022, American Chemical Society). | ||
Heating 8 to 80 °C supplies the necessary energy for the release of N2 and the formation of 9. This process follows a Staudinger pathway involving a four-membered ring transition state. The release of dinitrogen from 8 ultimately yields product 9 (Fig. 7). During the reaction of 7 with benzophenone, a selectively formed seven-membered ring product, 10, was obtained through a formal 1,4-addition of benzophenone. This highlights the characteristic of moderated and controlled reactivity of the Si–Si fragment within complex 7. In contrast, previous reports on the reaction of disilylene(I) with benzophenone resulted in selective C–O cleavage, yielding a cyclodisiloxane.34
In 2022, Khan and co-workers reported a new mixed donor ligand (11) (Scheme 4) and synthesized its M(II) complexes (M = Fe(II), Co(II), and Ni(II)). The authors also studied the electrochemical, optical, and magnetic properties of these complexes.35 The reaction of 11 with FeBr2 resulted in the formation of a four-coordinate silylene–Fe(II) complex 12 (Scheme 4). The molecular structure of 12 confirmed the distorted tetrahedral geometry around the Fe center, suggesting its paramagnetic nature (Fig. 8). The crystallization of compound 12 in the ferroelectric active space group Pna21, suggests a new application of silylene transition metal complexes. The magnetic property of complex 12 was studied using SQUEED magnetometry. At room temperature (300 K), complex 12 displayed paramagnetic behavior with an increasing linear trend of magnetization with magnetic field strength. In comparison, at lower temperatures (5 K), it showed soft magnetic behavior with S-shaped isothermal magnetization (M–H) curves and negligible coercivity (Hc) and remanence (Mr) magnetization values. The low coercivity and remanence values at lower temperatures suggest that complex 12 possesses properties of soft magnetic materials, making it suitable for electronic devices and magnetic data storage systems. The cyclic voltammogram (CV) studies of 12 also indicated its electroactive nature, with a reversible redox peak at Epa = 506 mV, corresponding to the Fe(I) → Fe(II) transition, and its corresponding reduction peak appeared at Epc = 436 mV. Another reduction peak at Epc = −1250 mV was attributed to Fe(I) → Fe(0), while the oxidation peak occurs at a more positive potential, Epa = 1644 mV, signifying the Fe(II) → Fe(III) transition.
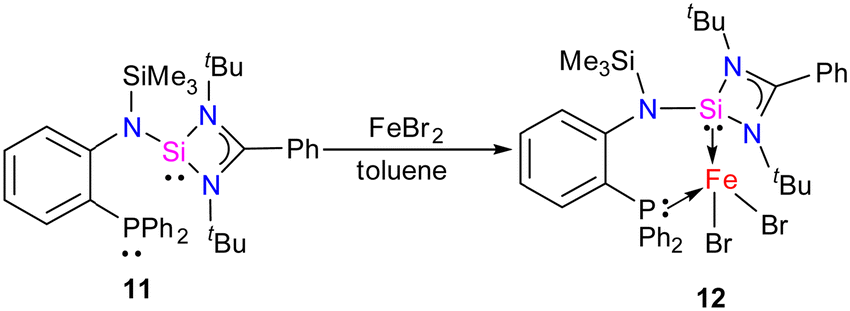 | ||
| Scheme 4 Synthesis of 12. | ||
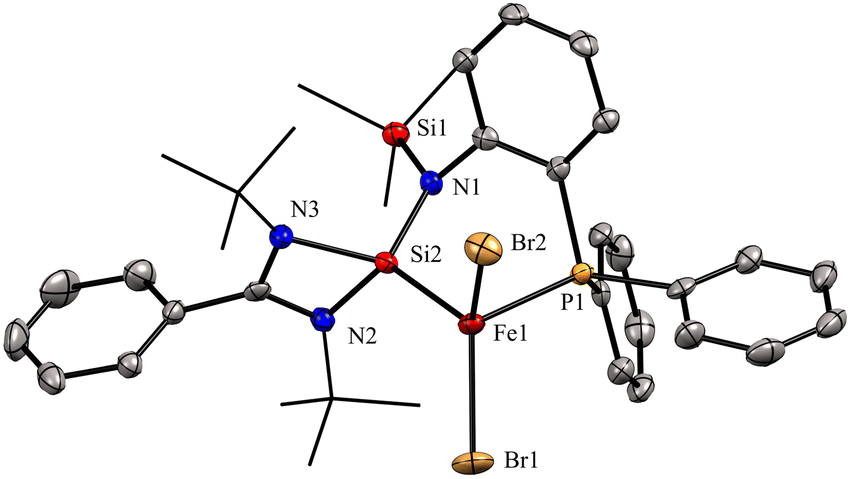 | ||
| Fig. 8 Molecular structure of 12 showing distorted tetrahedral geometry around Fe center. Reproduced from ref. 35 Copyright©2022, American Chemical Society. | ||
The effective magnetic moment of 12 was found to be 5.3 BM (linear fit) and 5.5 BM (Langevin fit), corresponding to four unpaired electrons. Consequently, it can be inferred that 12 is a high spin complex.
In 2022, Rieger and co-workers studied the electronic and steric properties of phosphinimide–silylene–Fe(0) complexes. The authors judiciously trapped the in situ generated silylenes with an olefin to afford phosphinimide-substituted siliranes. The reaction of N-trimethylsilyl-phosphinimide with SiBr4 afforded N-tribromosilyl-phosphinimides, which on further reduction with potassium hypersilanide [KSi(TMS)3] in the presence of cyclohexene afforded the desired phosphinimide-substituted siliranes. The phosphinimide-substituted siliranes on treatment with Fe(CO)5 led to the formation of silylene-substituted Fe complexes 13–15 (Scheme 5).36 The reaction, facilitated by gentle heating to 40 °C, involves the opening of the silirane ring, leading to the generation of transient silylenes. The 31P NMR signals indicate the electronic property changes in complexes 13–15. The signals are downfield shifted, transitioning from 14.02 ppm for 13 to 14.33 ppm for 14 and 51.49 ppm for 15. This shift aligns with the steric trend observed on Tolman's map of the phosphines. Simultaneously, the electronic trend is reflected in the 29Si NMR spectra, where a downfield shift is observed for the central silicon atoms. Specifically, the shift progresses from the complex of tBu–silylene with a signal at 293.4 ppm to the complex of Me2Ph–silylene at 311.5 ppm and, finally, to Ph–silylene at 320.5 ppm. The IR spectra of complexes 13–15 reveal a correlation similar to what is observed for phosphines on Tolman's map: an increase in the donor strength of the phosphine ligand results in a decrease in the wavelength of the CO vibrations (Table 1). This observation suggests that the relative donor strength of phosphinimide-based silylenes can be predicted by knowing the position of the utilized phosphine on Tolman's map.
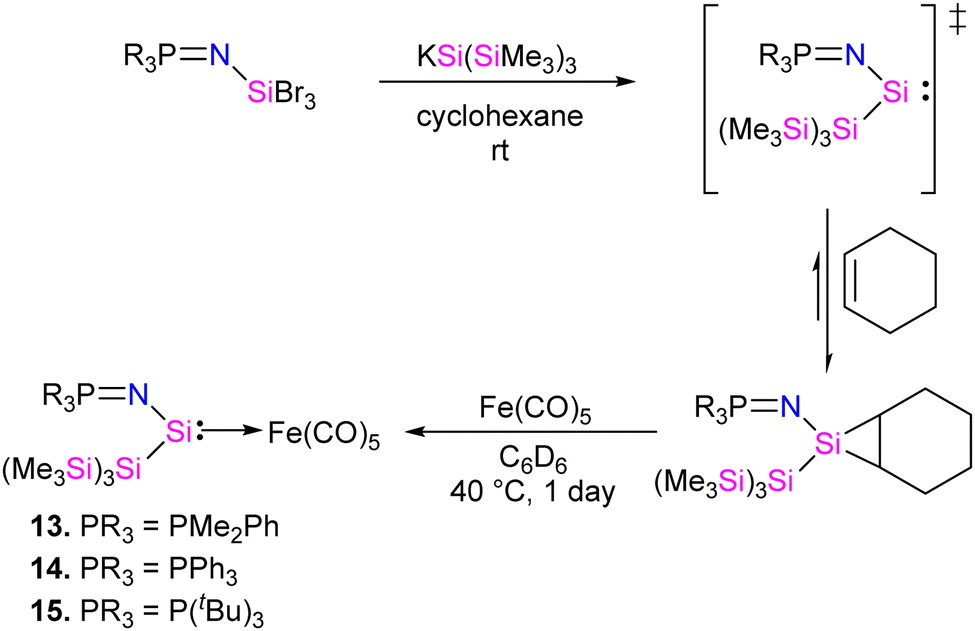 | ||
| Scheme 5 Synthesis of phosphinimide–silylene–Fe(0) complexes 13–15. | ||
| Fe(CO)4L | ν CO (cm−1) |
|---|---|
| Ph–silylene complex 14 | 2007, 1925, 1872 |
| Me2Ph–silylene complex 13 | 2003, 1921, 1869 |
| t Bu–silylene complex 15 | 1998, 1916, 1867 |
Dinitrogen functionalization, among various catalytic reactions involving silylene–Fe complexes, stands out as one of the most significant transformations. This is due to its pivotal role in numerous industrially relevant processes. Dinitrogen (N2) is the predominant gas in the Earth's atmosphere. However, its inert nature challenges its utilization as a nitrogen source in biosphere and industrial applications.37 Therefore, various approaches have been made to overcome this problem by reducing and functionalizing N2.38 In 2023, Li and co-workers designed and synthesized a new class of spacer-separated bis(silylene)–Fe(II) complexes and studied their catalytic reactivity in dinitrogen silylation reaction.39
The reaction of bis(pyrrol-2-yl)-methane derivatives with two equivalents of [PhC(NtBu)2SiCl] in the presence of a base LiN(SiMe3)2 resulted in the formation of bis(silylene) [SiCSi] pincer ligands 16–18. Which on further treatment with FeCl2(THF)1.5 in THF, resulting in the formation of tetra-coordinate bis(silylene) iron(II) chloride complexes 19–21 (Scheme 6).
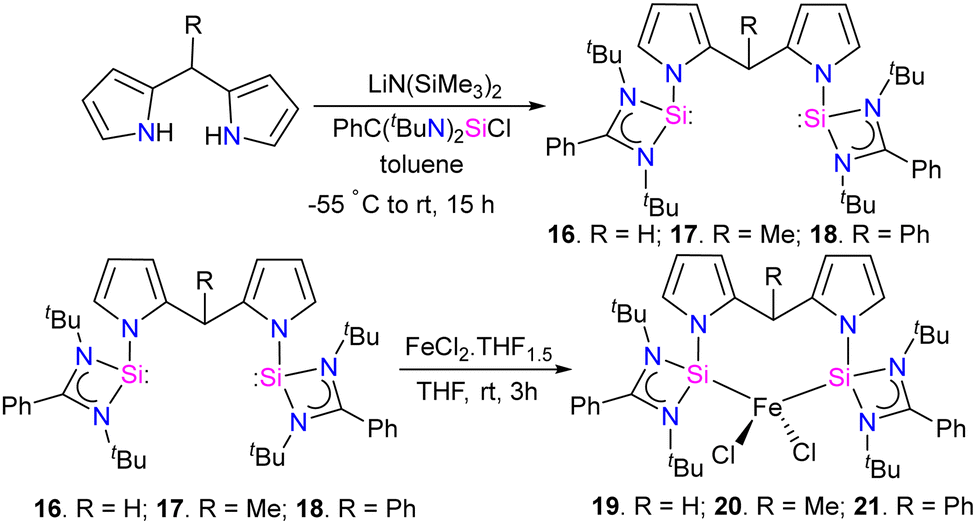 | ||
| Scheme 6 Synthesis of 16–18 and iron chloride complexes 19–21. | ||
The catalytic activity of complexes 19–21 was evaluated in a dinitrogen silylation reaction. Despite all three complexes exhibiting some degree of activity, it was found that the sterically bulky group on the central carbon had a positive impact. As a result, complex 21, showed the highest catalytic performance with an overall turnover number of 746. Notably, this represents the highest reported TON value for dinitrogen silylation among all silylene transition-metal catalysts. Furthermore, the authors found that the transient intermediates, pentacoordinated bis(dinitrogen) iron(0) complexes 22–24, function as the actual catalysts in N2 silylation reactions (Scheme 7).
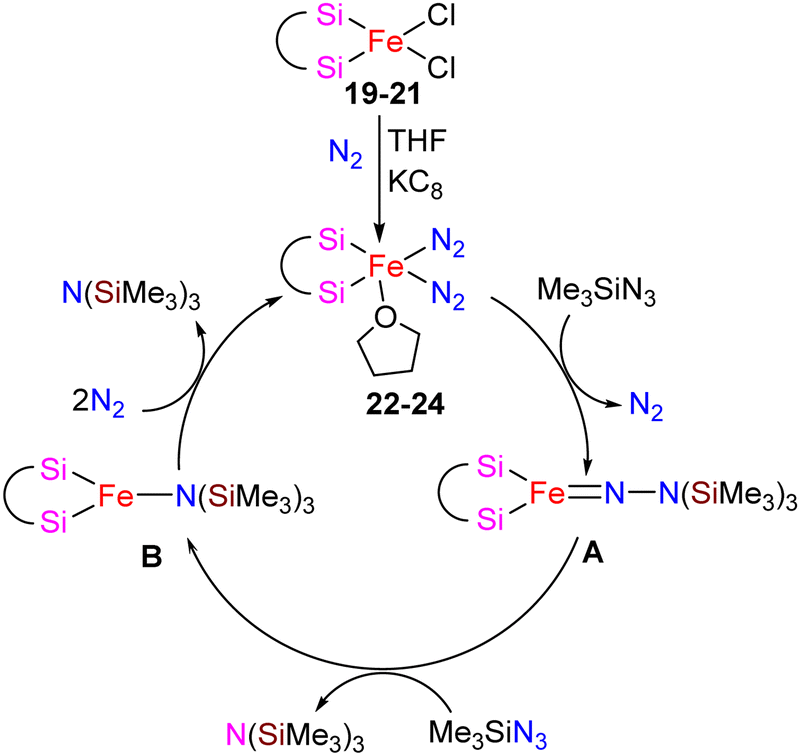 | ||
| Scheme 7 Proposed mechanism for the N2 silylation by Fe complexes 22–24. | ||
Bis-silylene synthesis with a reactive Si(I)–Si(I) bond represents a breakthrough in low-valent silicon chemistry. Recently, Roesky and co-workers synthesized a unique bis-silylene bridged by Si(IV) center.40 A 2:1 molar reaction of LSiCl (L = Ph(tBuN)2) with Ph2SiCl2 in the presence of 4 equivalents of KC8 led to the formation of unique bis-silylene 25. The 29Si NMR spectrum of 25 showed two resonances at −29.2 ppm and 59.9 ppm, corresponding to Si(IV) and Si(II) atoms, respectively. The Si(II)–Si(IV)–Si(II) bonding arrangement in the bis-silylene 25 is unique, with Si(II)–Si(IV) bond lengths of 2.4212(8) and 2.4157(7) Å. Further, the authors also explored the coordination ability of 25 with Fe(0) precursor. The reaction of 25 with two equivalents of Fe(CO)5 resulted in the formation of a dinuclear Fe(0) complex 26 (Scheme 8). The 29Si NMR spectrum of 26 showed resonances at −29.6 ppm and 113.0 ppm due to Si(IV) and Si(II) → Fe(CO)4 centers, respectively. As can be seen from the 29Si NMR chemical shift values, the silylene atom in Si(II) → Fe(CO)4 becomes highly deshielded due to coordination to Fe(CO)4 center when compared to the same in 25. The Si(II)–Si(IV)–Si(II) bond distances as well as the Si⋯Si distance is elongated upon coordination with Fe(0) (Fig. 9).
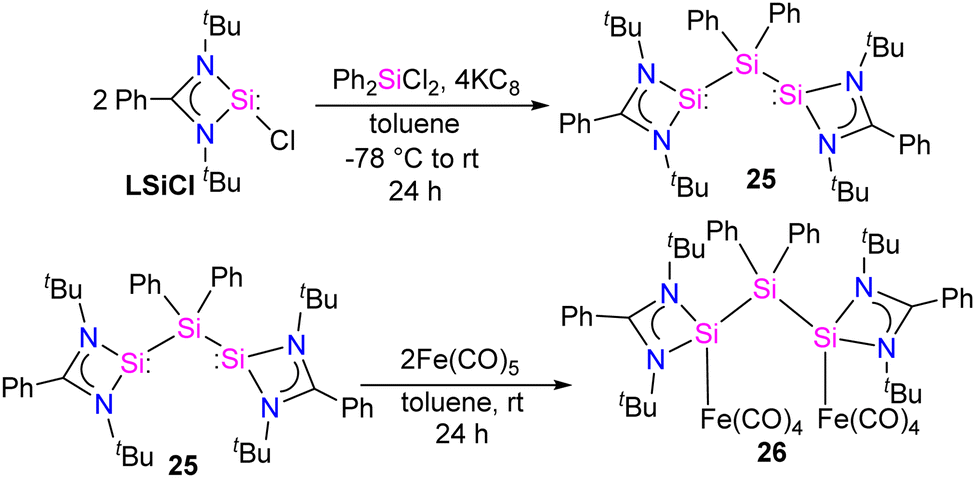 | ||
| Scheme 8 Synthesis of 25 and 26. | ||
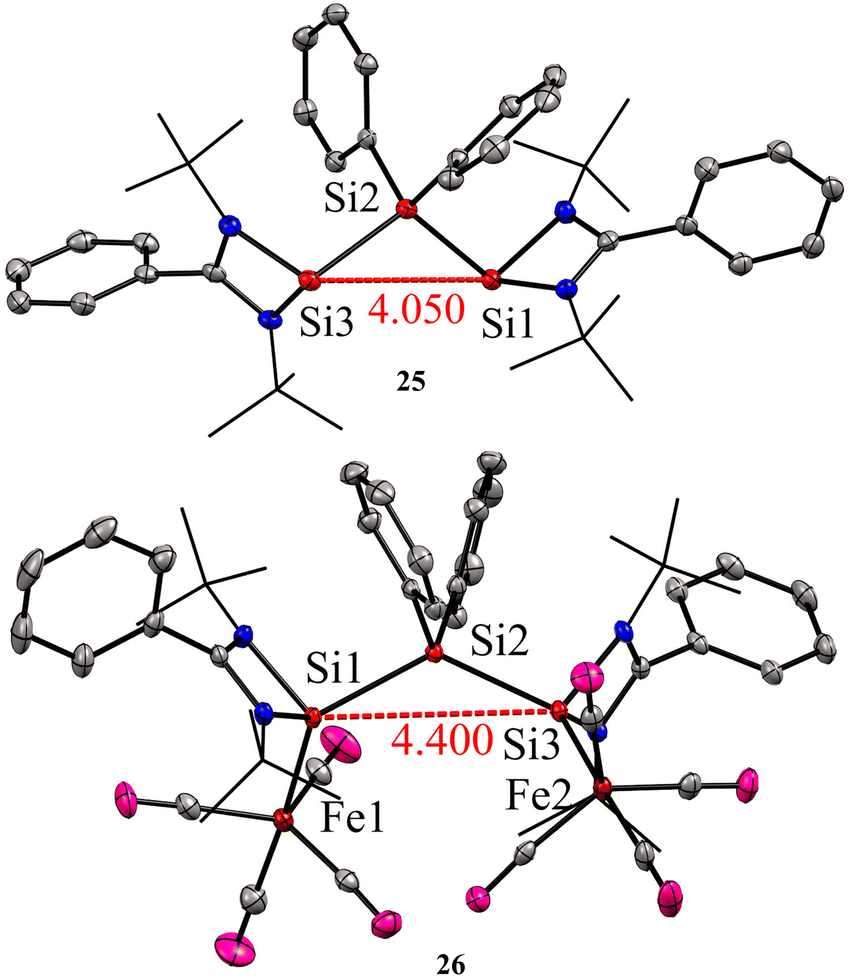 | ||
| Fig. 9 Molecular structures of 25 and 26 showing Si⋯Si distance is elongated upon coordination to Fe. Reproduced from ref. 40 Copyright©2023 Wiley-VCH GmbH. | ||
The IR spectrum of 26 showed frequency for CO at 1893 cm−1, which is slightly lower than the same in similar amidinate stabilized Fe(0) complexes, suggesting the electron-rich nature of 25.24 The authors have performed extensive DFT calculations to understand the nature of these Si–Si bonds. The Si–Si bonds in 25–26 are of an electron-sharing type, as suggested by NBO (natural bond orbital) and EDA-NOCV (energy decomposition analysis orbital for chemical valence) investigations.
Aminosilylene, comprising reactive –NH and Si(II) centre next to each other, is a versatile compound. Very recently, Roesky and co-workers used the concept of steric protection of the NH group to produce aminosilylene Ar*NHSi(PhC(NtBu)2) (27) (Ar* = 2,6-dibenzhydryl-4-methylphenyl) in its free form and studied its reactivity with Ru and Fe metal precursors.41 The reaction of [Li{NH(Ar*)}] with [(PhC(NtBu)2SiCl)], resulted in the formation of aminosilylene 27. The 29Si{1H} NMR spectra of 27 displayed a singlet at −3.4 ppm, akin to R2NSi(amidinate) (R = Cy, iPr).42 The 1H NMR spectrum of 27 exhibits a characteristic –NH proton singlet at 4.21, confirming compound formation.
While phosphine and nitrogen donor ligands are common in tethered Ru complexes, silylene ligands are seldom utilized in such systems.26 Treatments of 27 with [Ru(η6-p-cymene)Cl2]2 afforded a η6-arene tethered complex [RuCl2{Ar*NHSi(PhC(tBuN)2)-κ1-Si-η6-arene}] (28), whereas with the Fe(CO)5 precursor a Fe(0) complex [Fe(CO)4{Ar*NHSi(PhC(tBuN)2)-κ1-Si}] (29) was obtained (Scheme 9). The molecular structure of complex 28 confirmed the presence of a rare η6-arene tethered complex, whereas complex 29 exhibited a trigonal–bipyramidal geometry around the Fe atom, with ligand 27 occupying one of the apical positions to reduce steric hindrance, and four CO groups occupying the other positions (Fig. 10). In the case of 28, the HOMO is centered around the {RuCl2} group, wherein the Ru(dxy) orbital interacts with both Cl(3py) ions in a π* type manner. However, the LUMO is centered on the {RuCl2Ph} group, where Ru(dx2−y2) interacts with the Ph(π) and Cl(3ps) orbitals. Because of the interaction between Fe(dx2−y2) and CO(π*) orbitals, the HOMO for 29 is centered at {Fe(CO)4}. However, the LUMO is centered on the substituted silylene group and is of the π* type.
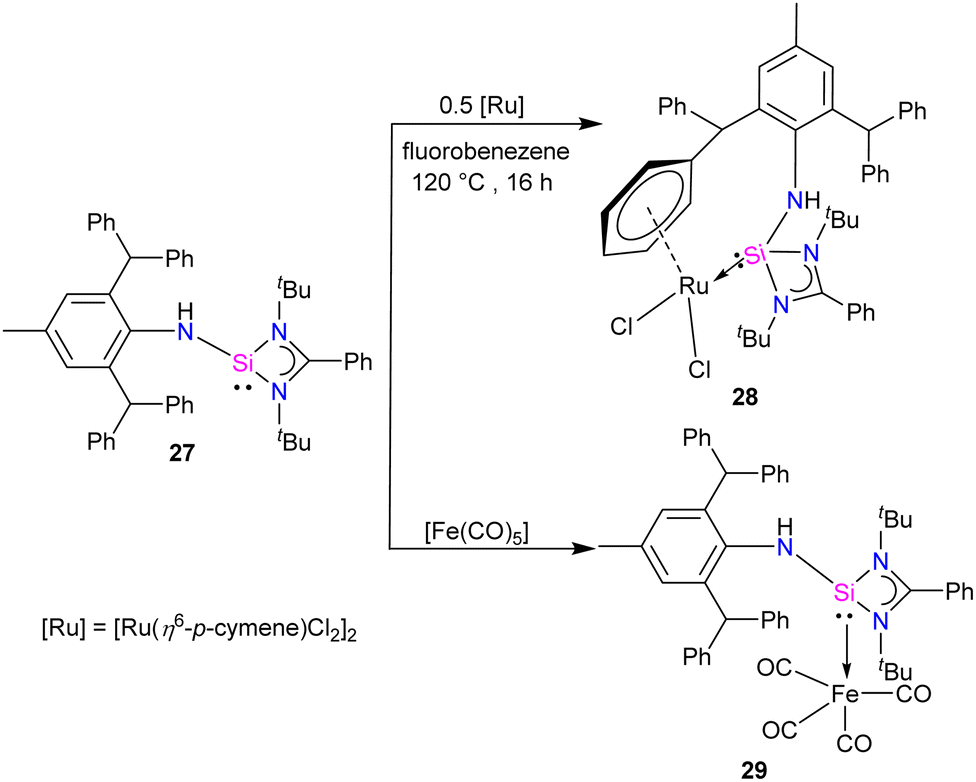 | ||
| Scheme 9 Synthesis of 28 and 29. | ||
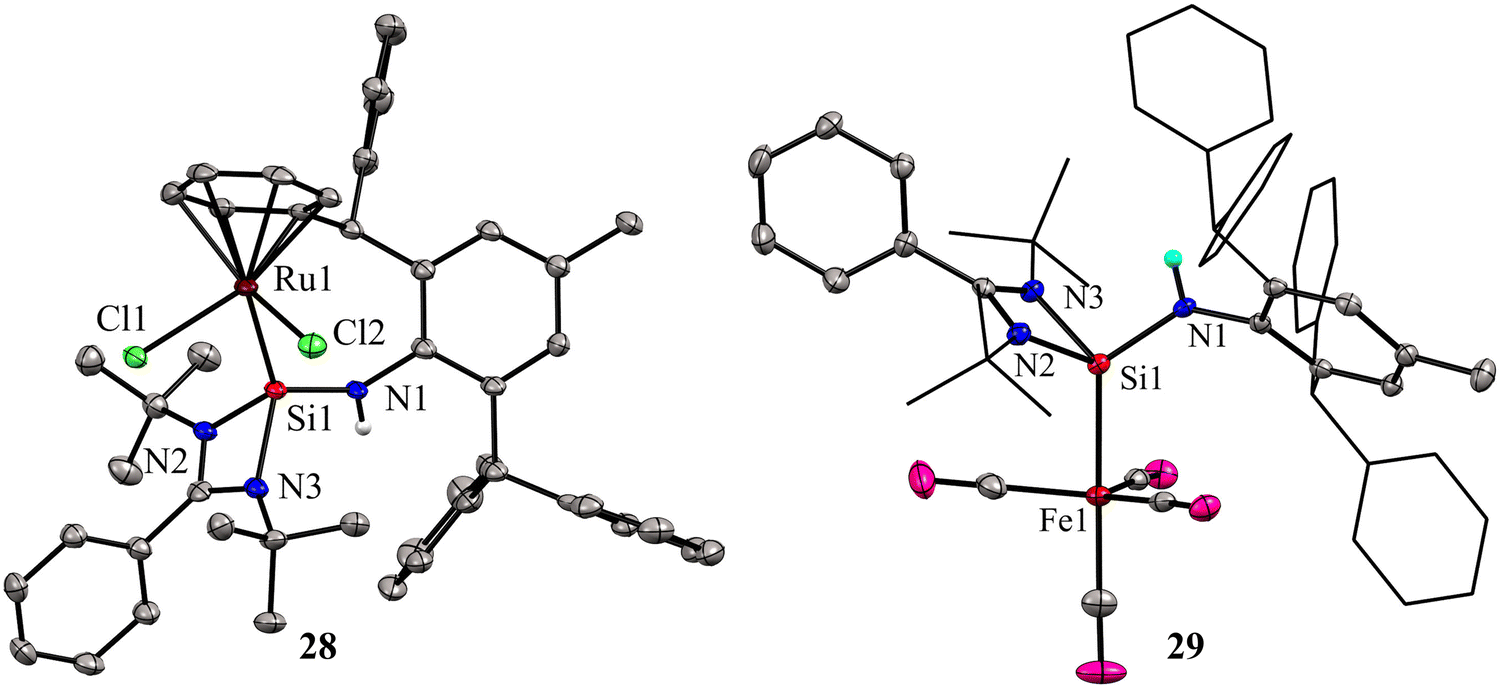 | ||
| Fig. 10 Molecular structures of 28 and 29. For simplicity, the phenyl and tBu groups are represented in the wire and sticks model. Reproduced from ref. 41 Copyright©2023 Wiley-VCH GmbH. | ||
In 2024, the same group utilized a short-bite bis(NHSi) ligand to synthesize a unique bimetallic Fe complex with a Fe–Fe bond distance of 2.6892(13) Å.43 The short-bite bis(NHSi) 30 was synthesized by the treatment of dilithiated amide ArNLi2 (Ar = p-toluidine) with [(PhC(NtBu)2SiCl]) in 1:2 molar ratios. The reaction of 30 with Fe(CO)5 in a 1:2 molar ratios afforded a unique bimetallic complex 31 featuring an intriguing five-membered (N–Si–Fe–Fe–Si) ring (Scheme 10). In complex 31, the ligand bite angle is expanded from 109.33(8)° to 118.2(2)° to accommodate two Fe atoms, and the ligand acts as a μ-bridging A-frame ligand to afford an interesting binuclear complex with Fe–Fe bond (Fig. 11). The 29Si NMR spectrum of 31 showed a singlet resonance at 36.5 ppm, which is considerably downfield shifted (49 ppm) compared to the free ligand 30 (−12.2 ppm), and the IR spectrum showed two types of CO stretching frequencies for the terminal and bridging CO groups in 2056 and 1851 cm−1 respectively. DFT studies have examined the strong Si⋯Si contact in complex 31, suggesting that the interaction is strong due to a back donation from Fe(0) to Si(II) atoms.
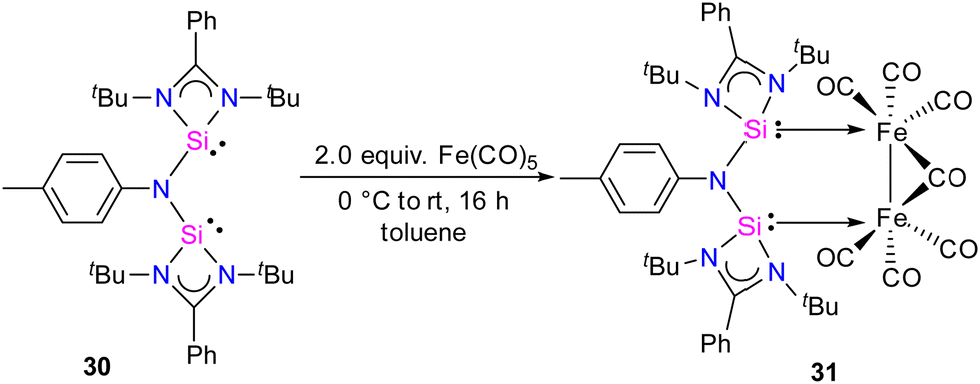 | ||
| Scheme 10 Synthesis of 31. | ||
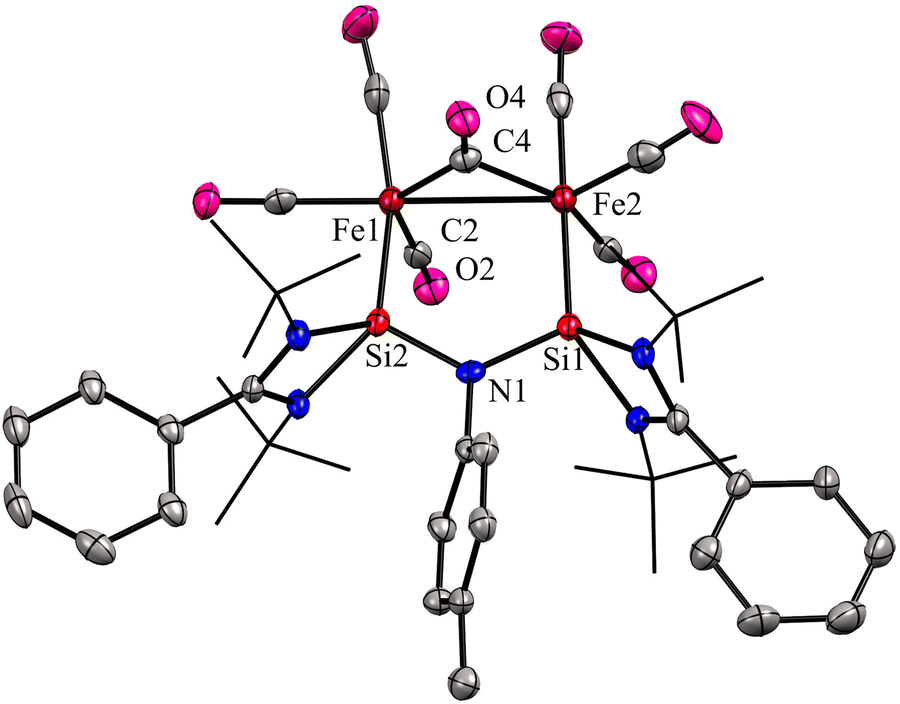 | ||
| Fig. 11 Molecular structure of 31. Reproduced from ref. 43 Copyright © 2023 Wiley-VCH GmbH. | ||
Co–silylene complexes
Hydrogenation of olefins is one of the most important transformations in organic synthesis, owing to its application in producing agrochemicals, pharmaceuticals, and commodity chemicals.44 Driess and co-workers recently reported the olefin hydrogenation catalyzed by NHSi, they utilized a bis(N-heterocyclic silylene)xanthene nickel(0) complex as an efficient precatalyst for the hydrogenation of olefins.45 However, the olefin hydrogenation with Co complexes of silylene ligands was not explored. The success achieved using NHSi ligands in various catalytic transformations and with the knowledge that low-valent cobalt complexes are known to serve as effective catalysts for the hydrogenation of olefins. Mo and co-workers hypothesized that N-heterocyclic imino substituted silylene owing to its strong σ-donor ability and the presence of a sterically demanding group in the NHSi backbone might stabilize low-valent Co(I) complex.The reaction of 32 with CoCl2 in the presence of reducing agent Na/Hg afforded a unique arene-tethered complex 33 (Scheme 11). On further treatment with Bu3BHK afforded the cobalt hydride complex 34.46 Both the complexes were diamagnetic and showed a highly deshielded singlet resonance in the 29Si NMR spectra at 35.27 and 40.83 ppm, respectively (−26.97 ppm for 32). The characteristic Co–H resonance for 34 in the 1H NMR spectrum was observed at −13.2 ppm, suggesting the Co-bound hydride ligand in 34. With the cobalt hydride complex 34, catalytic hydrogenation of styrene was investigated. The cobalt hydride complex 34 was found to be a very efficient catalyst for the olefin hydrogenation reactions under very mild conditions (5 mol% 34, 60 °C, 1 bar H2 gas). The reaction worked well even with 0.5% catalyst loading but required more time (24 h) for completion. No product formation was observed when only NHSi ligand was employed, hence suggesting the crucial role of Co center in the catalysis. Further, the authors also checked a variety of substituted olefins under the optimum conditions and found that the reaction worked well and afforded quantitative yields of hydrogenated products in all of the cases (Scheme 11). While the detailed mechanism was not probed, the authors hypothesized that the reaction starts with the insertion of Co–H to olefin to afford cobalt alkyl complex, which might undergo σ-bond metathesis with H2 gas to afford the hydrogenated product together with the regeneration of the catalyst 34.
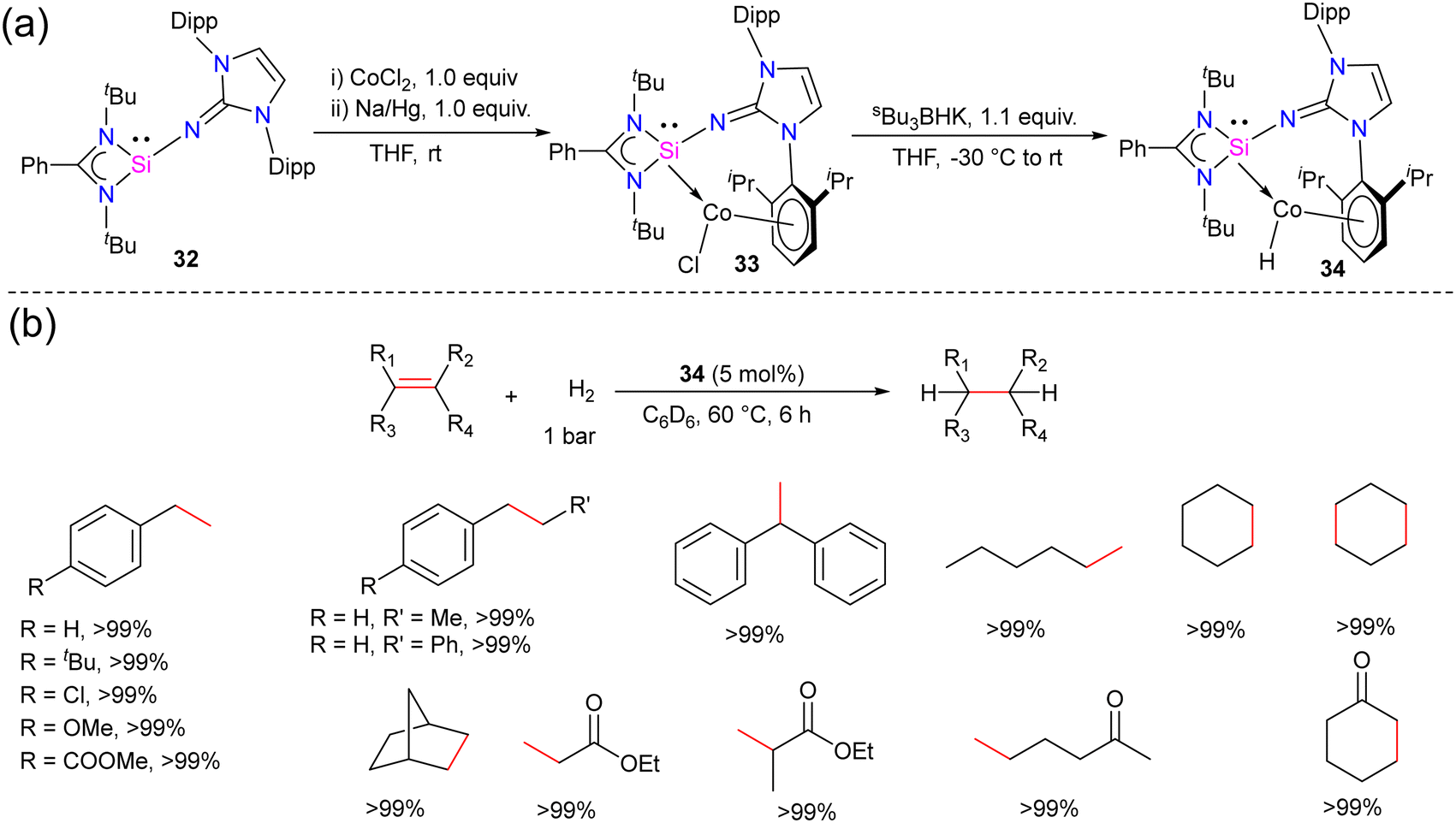 | ||
| Scheme 11 (a) Synthesis of NHSi stabilized Co(I) complexes 32–34. (b) Catalytic utility of 34 in olefin hydrogenation. | ||
To get insight into the impact of replacing phosphine with silylene ligands, Hinz and Li studied the hydrosilylation of carbonyl compounds catalyzed by Co(PMe3)3Cl and Co(LSi:)2(PMe3)2Cl (35) (LSi: = {PhC(NtBu)2}SiCl) complexes.47 The reaction of [(PhC(NtBu)2SiCl)] with Co(PMe3)3Cl in 2:1 molar ratio afforded the desired complex Co(LSi:)2(PMe3)2Cl (35) as red solid in excellent yield.48 The 29Si NMR spectrum of 35 showed a singlet resonance at 30.0 ppm which is slightly downfield shifted when compared to [(PhC(NtBu)2SiCl)] (14.6 ppm). The authors found that both complexes are active catalysts for the hydrosilylation of carbonyl compounds. However, the replacement of phosphine with silylene in Co(LSi:)2(PMe3)2Cl (35) proved beneficial, and the complex showed higher activity in the catalytic hydrosilylation of aldehydes. Under the optimized condition, this catalyst reduced a series of substituted aldehydes to corresponding alcohols with good to excellent yields (Scheme 12). In contrast, the Co(PMe3)3Cl complex was more active for ketone hydrosilylation. The authors also probed the mechanism for catalytic hydrosilylation reactions and found that the hydrosilylation of aldehydes catalyzed by Co(PMe3)3Cl proceeds via a mechanism different from that of the analogous reaction with complex 35. However, in the case of ketones, both complexes catalyze the reaction using the same mechanism.
 | ||
| Scheme 12 Hydrosilylation of carbonyl compounds by Co complex 35. | ||
Catalytic hydrogen isotope exchange (HIE) reactions play a pivotal role in synthesizing deuterated and tritiated molecules, indispensable in diverse fields such as pharmaceuticals and medicinal chemistry.49 Traditionally, the advancement of HIE methodologies utilizing transition metals has leaned heavily on precious metal catalysts, owing to their efficacy in activating C–H bonds.50 However, recent research has shifted its focus towards exploring first-row transition metal alternatives, which offer distinctive advantages in terms of selectivity compared to the more conventionally employed precious metals. Pioneering investigations by Chirik51 and De Ruiter52 have underscored the importance of diverse electronic properties in catalytic HIE reactions, particularly by utilizing iron complexes bearing electron-rich pincer ligands. Driess and co-workers demonstrated that the substitution of phosphine and nitrogen donors with N-heterocyclic silylenes can furnish electron-rich metal centers, thereby enhancing catalyst activity for C(sp2)–H borylation reactions.53 The incorporation of ligands containing N-heterocyclic silylenes has been identified as a means to enhance the electron richness of metal centers, thereby boosting improvements in HIE methodologies. Building upon this foundation, Chirik and co-workers employed their previously reported54 well-defined bis(silylene)pyridine cobalt(III) dihydride boryl, trans-[ptolSiNSi]Co(H)2BPin (ptolSiNSi = 2,6-[EtNSi(NtBu)2CAr]2C5H3N, ptol = 4-MeC6H4, Pin = pinacolato) (36) complex as precatalyst in HIE reactions involving arenes and heteroarenes using benzene-d6 as the deuterium source.55
The authors first tested the HIE reactions using 1-(H)2BPin (36) complex and D2 gas as a deuterium source, which resulted in only moderate deuterium incorporation. Stoichiometric studies with H2 gas afforded a mixture of [ptolSiNSi(H)2][Co(H)2BPin] (37) and [ptolSiNSi(H)][CoH2(H2)] (38) complexes formation by irreversible modification of the pincer ligand through H2 addition, leading to catalyst deactivation (Scheme 13). The reactivity of 1-(H)2BPin (36) in C6D6 was explored to evaluate benzene-d6 as a deuterium source. Heating benzene-d6 solution of 36 at 60 °C resulted in deuterium incorporation into cobalt hydrides and the 4-position of pyridine, demonstrating C6D6 as a potential source for catalytic HIE reaction. The standard catalytic conditions comprised 1 mol% of 1-(H)2Bpin (36) in a 0.25 M substrate solution dissolved in benzene-d6, at 80 °C. This protocol worked well for a diverse range of substrates and provided good to excellent deuterium incorporation in the majority of the cases by facilitating C–H activation at sterically hindered sites (Scheme 14).
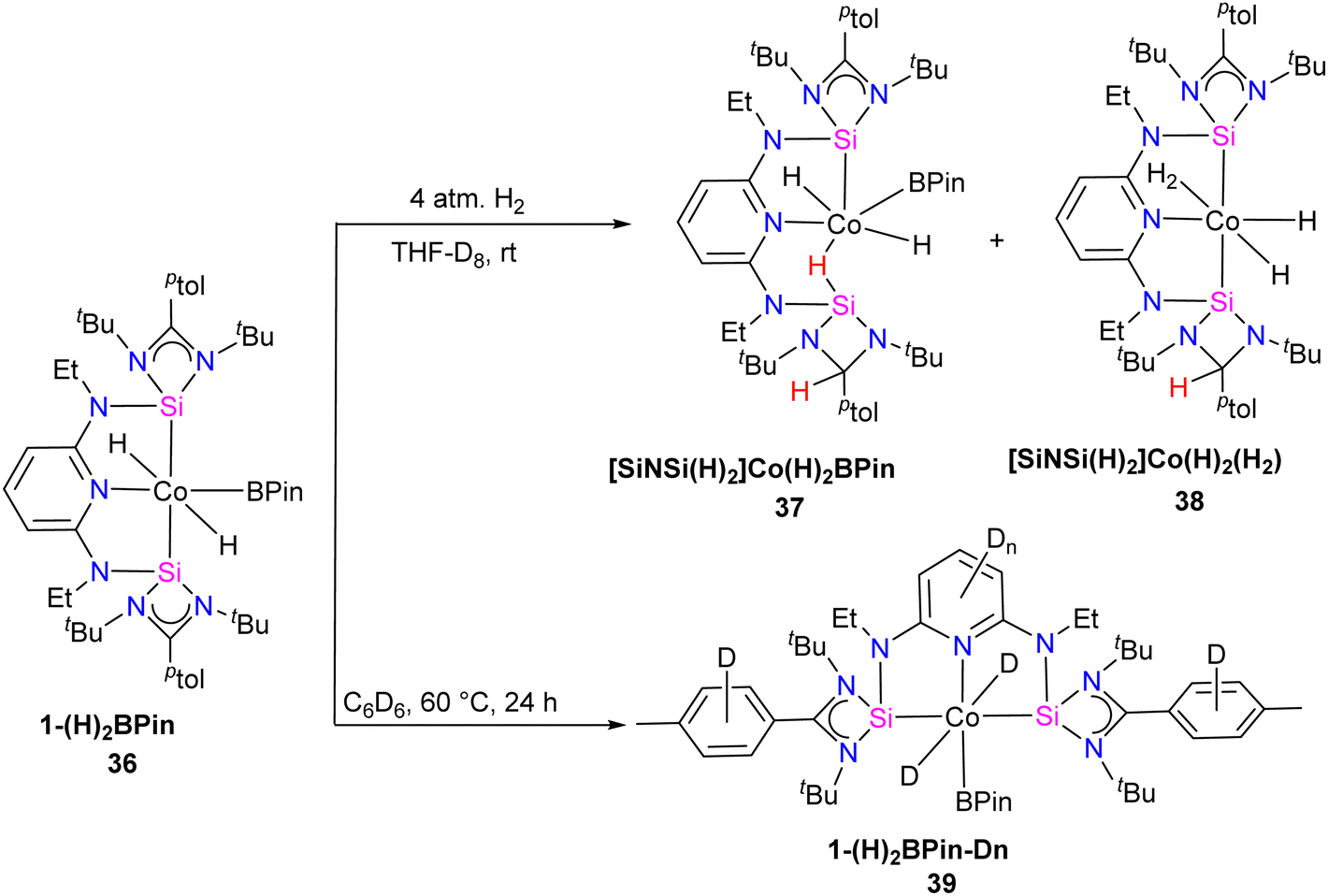 | ||
| Scheme 13 Synthesis of complexes 37 and 38 (pincer modification by H2 addition) and 39 (deuterium incorporation evaluation). | ||
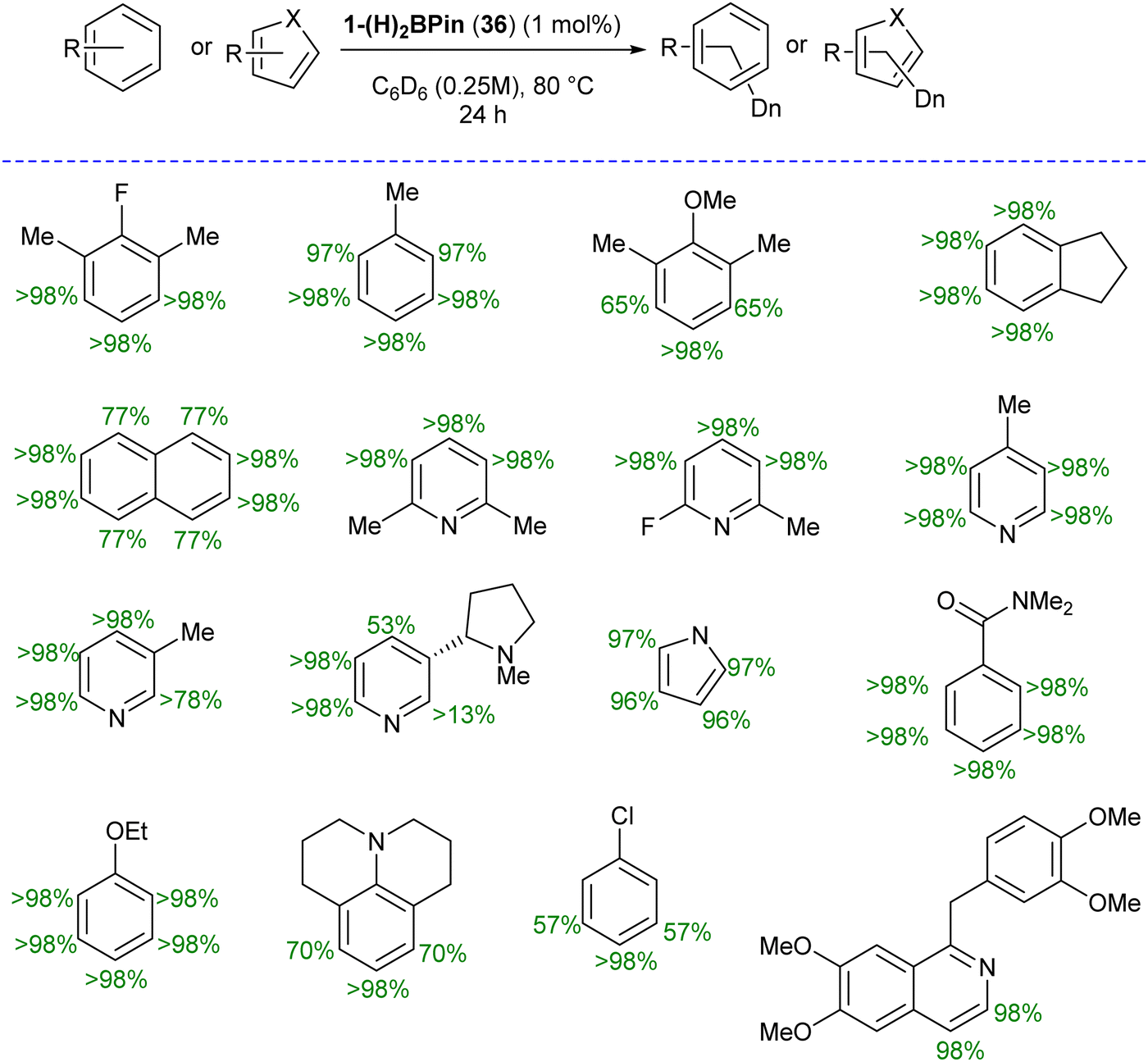 | ||
| Scheme 14 Substrate scope for HIE reaction using 36. | ||
The method was also compatible with aryl halides, favouring chemo-selective C(sp2)–H over C(sp2)–X (X = Cl, Br) bond activation. NMR monitoring reveals cobalt(III) resting states and inhibition by HBPin addition. Studies on precatalyst activation support bis(hydride)aryl cobalt intermediates in the catalytic HIE process. Mechanistic insights lead to an optimized protocol using [ptolSiNSi]Co(H)3·NaBHEt3 as the precatalyst, enhancing isotopic incorporation. A proposed mechanism for the catalytic HIE reaction is depicted in Scheme 15. First the 1-(H)2BPin complex 36 loses HBPin to generate a cobalt(I) hydride complex, subsequently the cobalt(I) hydride reacts with benzene-d6 to form a cobalt(I) deuteride that transfers deuterium to the substrate (fluoro benzene) and regenerate the cobalt (I) hydride. Post-catalytic turnover, HBPin may react with cobalt(I) hydride, forming the dihydride boryl resting state 36.
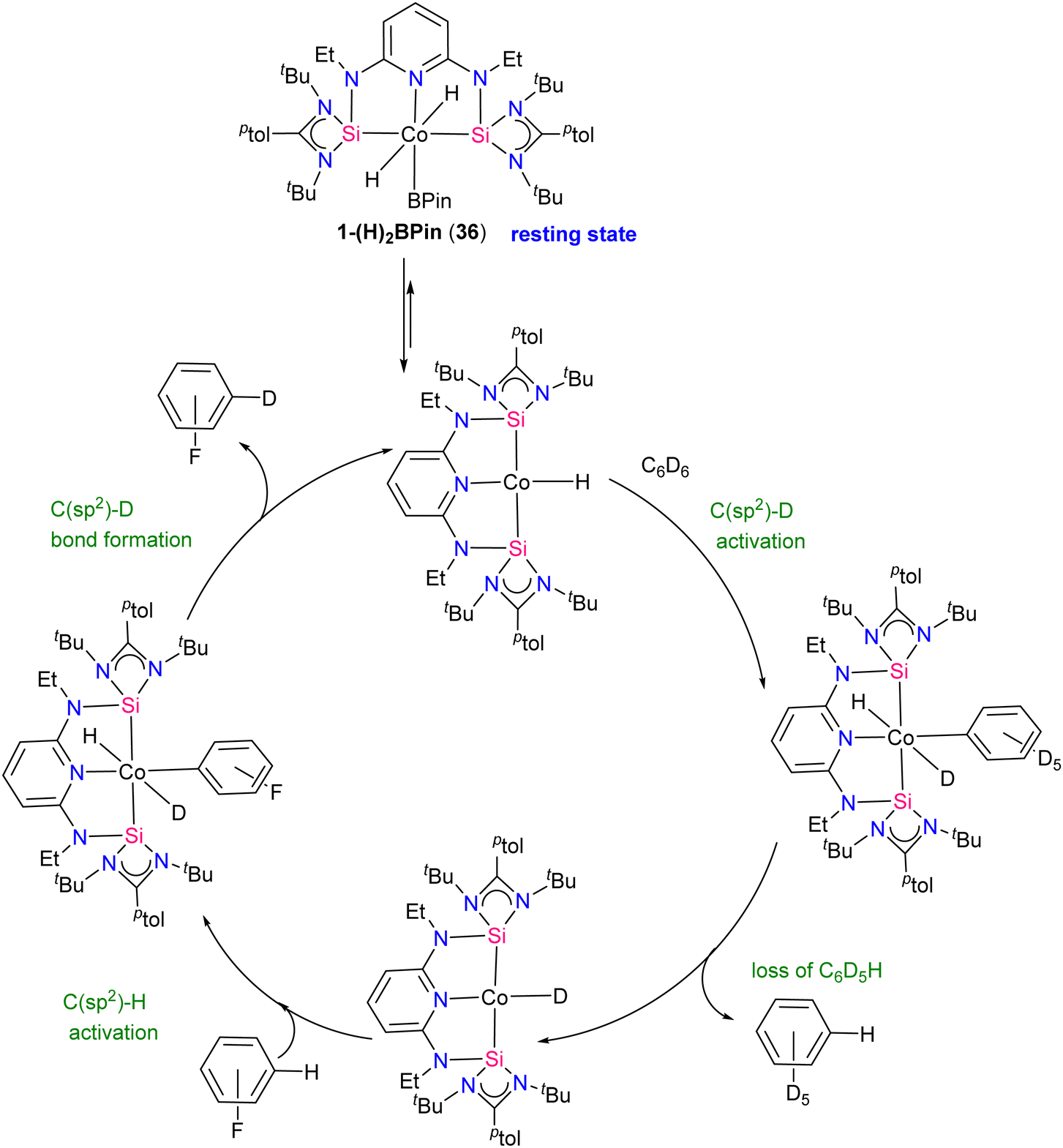 | ||
| Scheme 15 Proposed pathway for the HIE reaction showing the experimentally observed resting state. | ||
Mn complexes of silylenes
The chelating bis(silylene) ligands stabilize main-group elements and transition metals in low-valent states due to their strong σ-donor attributes.22b However, examples of Mn(0) complexes stabilized by silylenes are rare. An earlier attempt from our group to obtain a Mn(0) complex stabilized by silylene (Si → Mn(0)) was unsuccessful. It led to a disproportionation reaction, forming a silylene–Mn(I) complex (XXVI).27 The 17 valence electron (VE) Mn(0) compound Mn(CO)5 has only been achieved in low-temperature matrices.56 Encouraged by the success achieved using SiNSi pincer ligand derived from diaminopyridine backbone in the isolation of Fe(0) complexes and their utility in carbonyl hydrosilylation reactions.57 The authors utilized the SiNSi pincer ligand to synthesize Mn(0) complexes and studied its reactivity and catalytic properties.58 The reaction of SiNSi ligand with one equivalent of MnX2 (X = Cl, Br) resulted in four-coordinate Mn(II) complexes (40–41), akin to Mn(II) complexes of carbenes. However, the Mn(II) complexes of PNP59 and NNN60 pincer ligands generally adopt a five-coordinate coordination environment around Mn. This stark difference in the coordination environment around the Mn center was supposed to be due to the stronger σ-donor nature of the bis(silylene) arms, which forces the Mn center to adopt tetrahedral coordination. The reduction reactions of Mn(II) complexes 40 and 41 using KC8 without any supporting ligand did not work. The presence of a supporting ligand and the sequence of its addition to the reaction strongly affected the reaction yield. The reduction of 40 and 41 with KC8 followed by the addition of dmpe (dmpe = 1,2-bis-dimethyl phosphinoethane) resulted in the formation of an unusual Mn–H complex 42 (Scheme 16). The reaction was proposed to proceed via the formation of Mn(0) complex as an intermediate. Nevertheless, altering the order of dmpe addition by introducing it before the addition of KC8 resulted in the successful production of the intended Mn(0) complex 43. Further, the reaction of 43 with CO gas under ambient reaction conditions led to the replacement of supporting ligand dmpe by three CO groups, forming a unique 17-valence electron Mn(0) complex 44. The IR spectrum of 44 showed three CO stretching vibrations at 1844, 1811 and 1716 cm−1, which are shifted to a lower frequency compared to known Mn(0) complex [Mn(CO)3(CNArDipp2)2] (ArDipp2 = 2,6-(2,6-(iPr)2C6H3)2C6H3),61 suggesting strong backdonation from Mn to CO group in 44. Further, the Mn(0) complex 43 was utilized as a pre(catalyst) for the regioselective hydrogenation of N-heteroarenes. Complex 43 was superior to the other complexes, and under optimized conditions, a series of N-heteroarene were regioselectively hydrogenated with this practical catalyst (Scheme 16).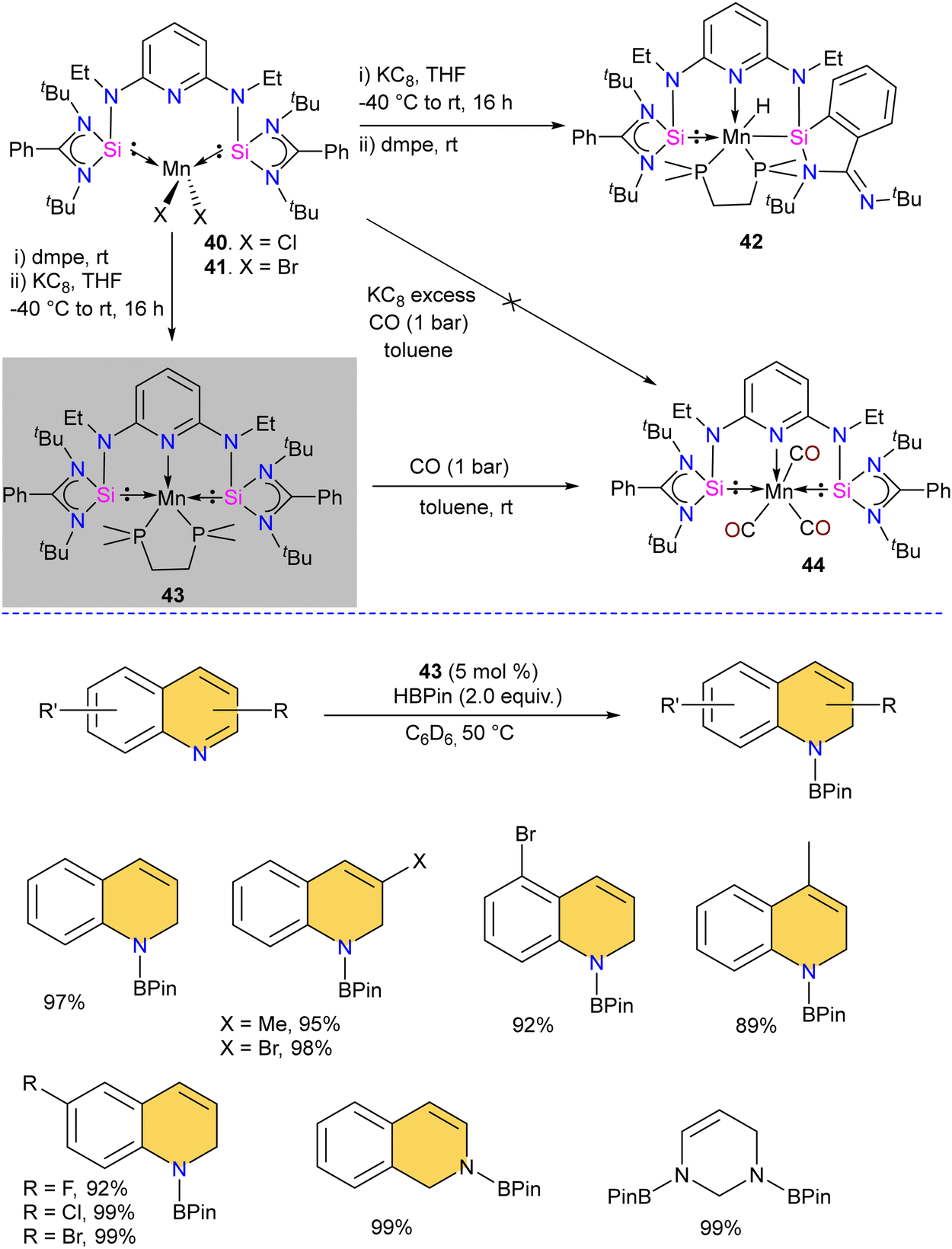 | ||
| Scheme 16 Synthesis of 40–44 and catalytic N-heteroarene hydrogenation using 43. | ||
Given that silylenes are better donors than phosphines and carbenes, Arevalo and co-workers utilized an Mn(SiNSi)Cl2 complex for the C(sp)–H borylation of terminal alkynes.62 The authors tested a series of Mn complexes stabilized by different donors in the catalytic C–H borylation of 4-fluorophenylacetylene with HBPin (Pin = pinacolate). Silylene stabilized Mn(SiNSi)Cl2 complex 40, was found to be the best catalyst and provided a good yield of alkynylboronate ester. Control experiments (with only ligand and with only MnCl2) suggested a crucial role of well-defined Mn(SiNSi)Cl2 complex in catalysis.
The authors varied the reaction condition to find the optimum conditions, and it was found at 80 °C, with 5 mol% of Mn(SiNSi)Cl240 and 2.5 equiv. of HBPin is the best condition for this reaction. Under optimum conditions, alkynes with electron-withdrawing andelectron-releasing substituents were successfully borylated in good to excellent yield (Scheme 17). Further, the authors have done stoichiometric studies to get insight into the mechanistic pathways. The reaction of 40 with HBPin was crucial for generating a catalytically active complex, and the authors hypothesized that the catalyst 40 enters the catalytic cycle after reaction with HBPin.
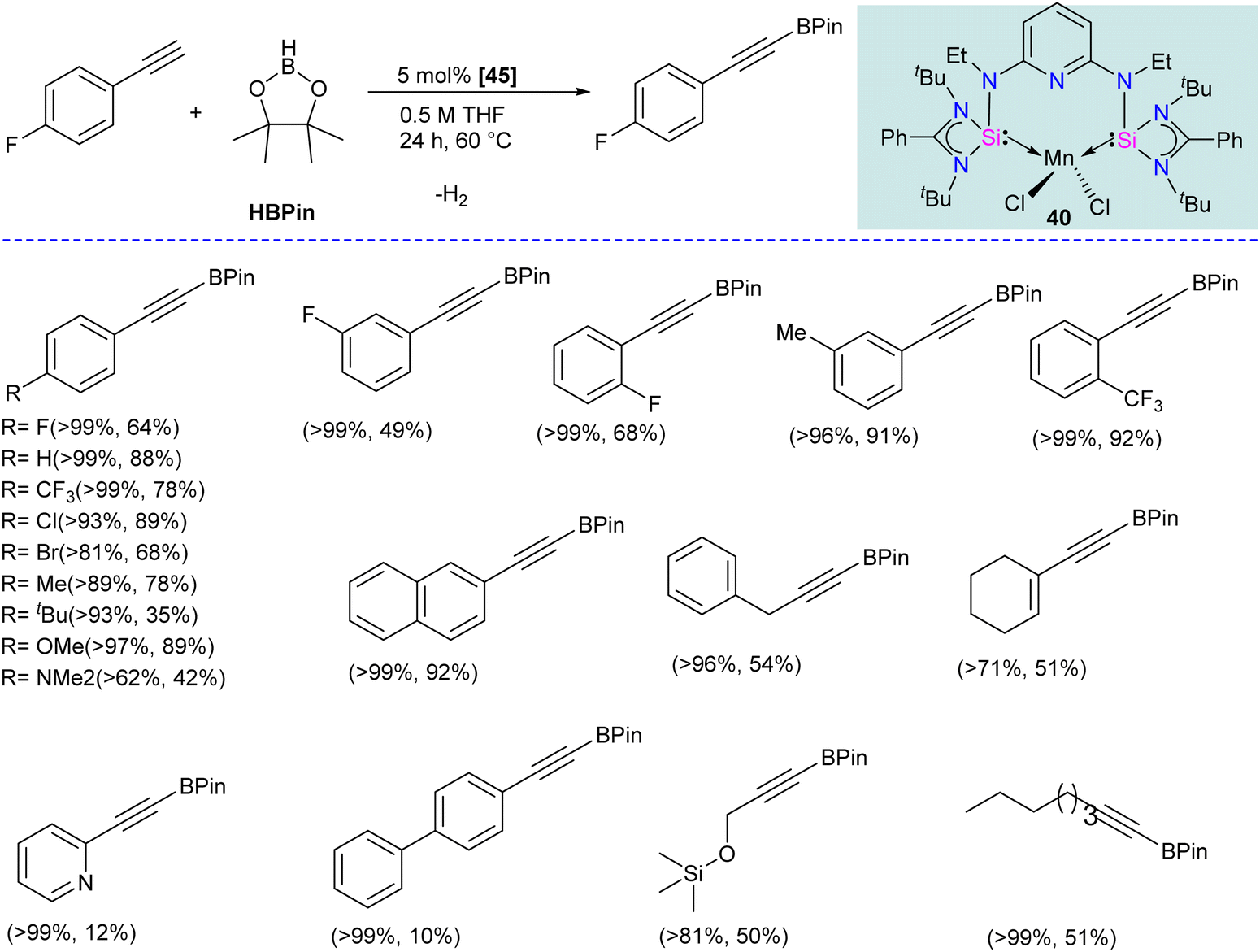 | ||
| Scheme 17 The catalytic utility of SiNSi–MnCl2 complex 40 in C(sp)–H borylation reactions. | ||
Compounds featuring Ni–Si (silylenes) bond
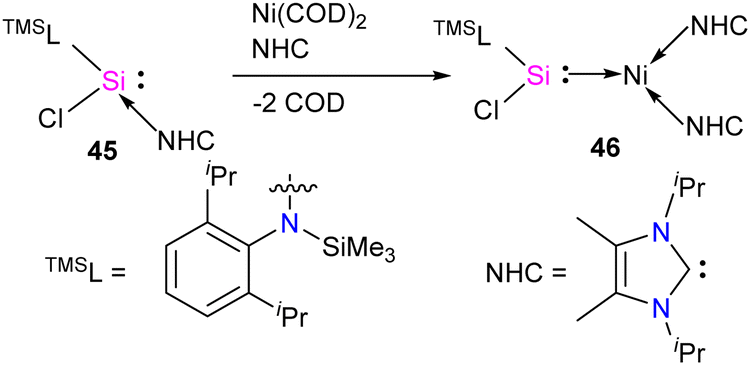
The first silylene–nickel complex [Ni(CO)2(tBu2NHSi)2] was reported by West in 1994, which was trigonal planar at the Ni center.63 Subsequently, Lappert and co-workers reported a homoleptic tetrahedral complex Ni{L}4 {L = Si[(NCH2tBu)2C6H4-1,2]} in 1998, by the reaction of Si[(NCH2tBu)2C6H4-1,2] with Ni(COD)2.64 However, West silylene (tBu2NHSi) reaction with Ni(COD)2 only afforded a trigonal planar Ni(0) complex. Following these initial breakthroughs, several attempts have been made to synthesize and characterize new Ni complexes of silylenes and study their reactivity (Fig. 12), which are thoroughly discussed in a recent review by Li and co-workers.17b This class of compounds usually demonstrates excellent reactivity toward small molecules and mostly are known for their efficiency in small molecule activations.17c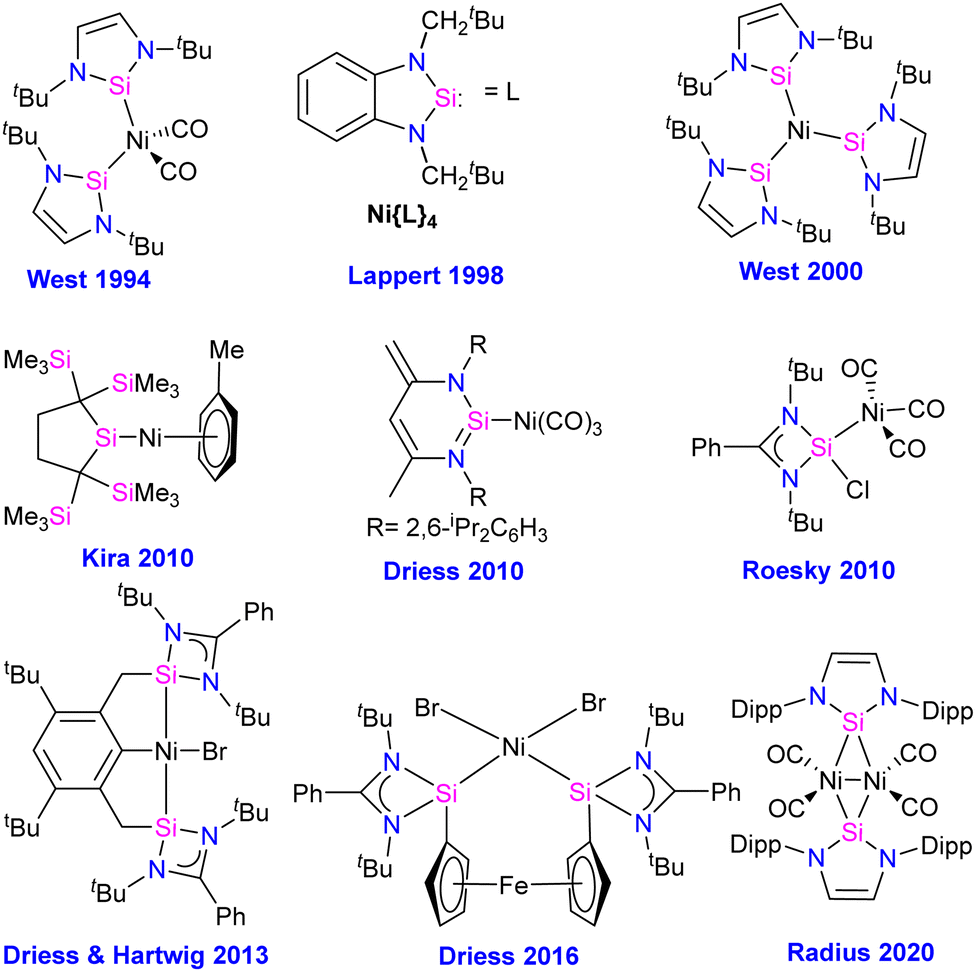 | ||
| Fig. 12 Selected examples of Ni complexes of silylenes. | ||
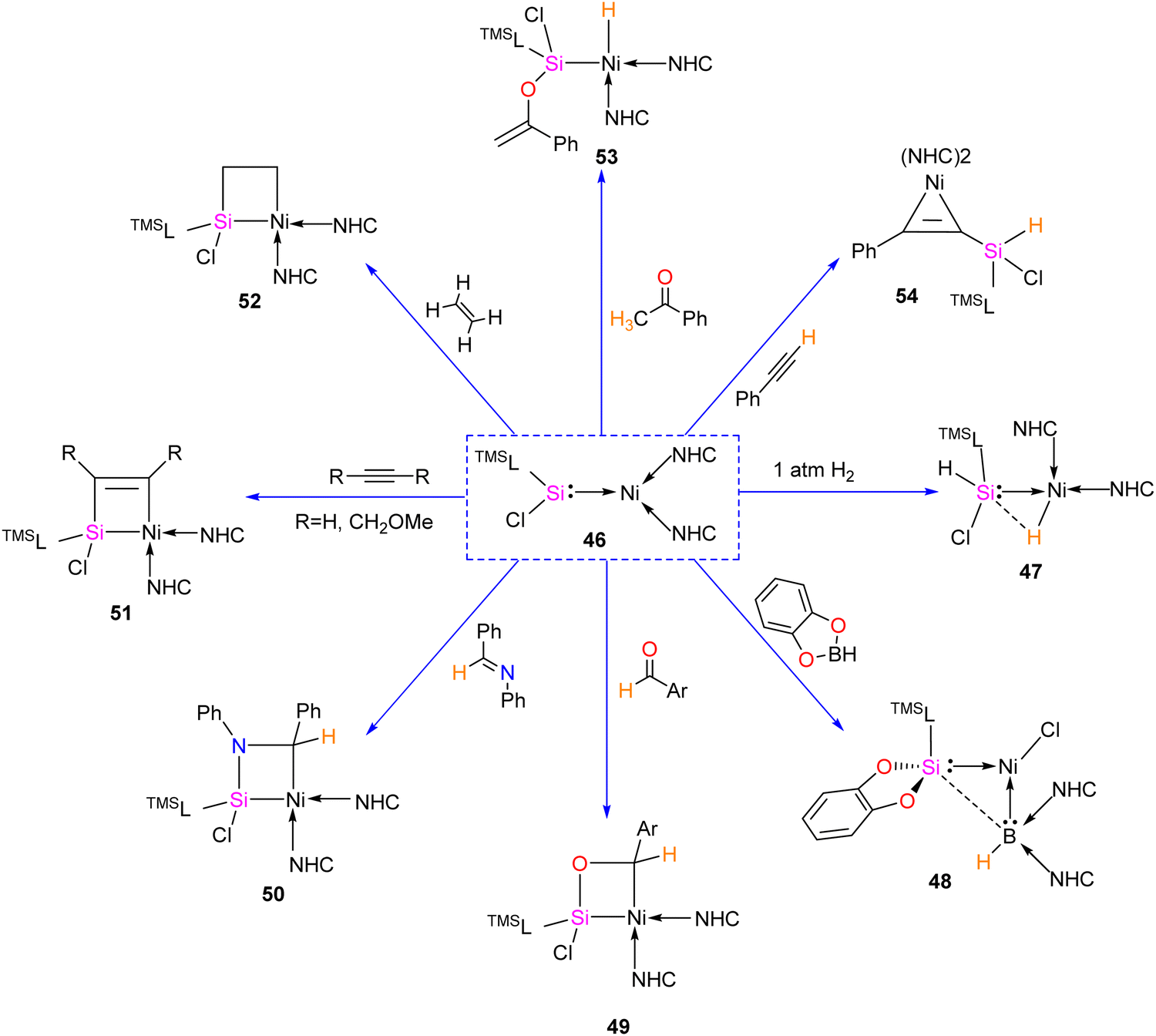
Most of the Ni complexes of silylenes contain 18 valence electrons. In 2017, Driess and co-workers synthesized a 16 valence electron (VE) silylene Ni(0) complex 46 by a silylene transfer reaction utilizing a readily accessible NHC-stabilized acyclic silylene (Scheme 18).65 This opened a new doorway in small-molecule activation chemistry, and the same group examined its reactivity with H2, catechol borane, and organic π-systems (Scheme 19).66 The reaction of 46 with H2 (1 atm.) resulted in the facial H2 activation and formation of the hydrido(silyl)nickel complex [(TMSL)ClSi(H)Ni(H)(NHC)2] (47). In contrast, catechol borane resulted in the reductive transformation of HBcat to a monovalent BH ligand and the formation of complex [cat(TMSL)Si(Cl)Ni ←:BH(NHC)2] (48).
 | ||
| Scheme 18 Synthesis of 16 valence electron (VE) Ni(0) complex. | ||
 | ||
| Scheme 19 The reactivity of 46 toward small molecules. | ||
The four-membered nickelasilacycle formation was observed in 49–52 when 46 was treated with unsaturated organic compounds.
The [2+2] cycloaddition reaction of unsaturated organic compounds with Si–Ni multiple bonds led to the formation of a four-membered nickelasilacycle 49–52. In the case of the reaction of 46 with acetophenone and phenylacetylene, both of which feature acidic C–H bonds, C–H bond activation occurred and resulted in the formation of 53 and 54, respectively. This indicates a low tolerance of 46 towards relatively acidic C–H moieties in these reactions. A noteworthy observation is that the addition of ethylene is reversible. However, when exposed to excess ethylene, the reaction undergoes a [2+2+2] cycloaddition, culminating in the activation of C(sp2)–H bonds in 56 (Scheme 20). The reaction is proposed to proceed via formation of 1-nickela-4-sila-cyclohexane intermediate 55. Although complex 55 was highly unstable, it could be isolated by the reaction of 46 with excess ethylene at −30 °C. Complex 55 on Ni mediated β-hydride elimination/reductive elimination afford complex 56. Further, complex 46 on treatment with an excess of 2-butyne proceed through the elimination of silole 57a.
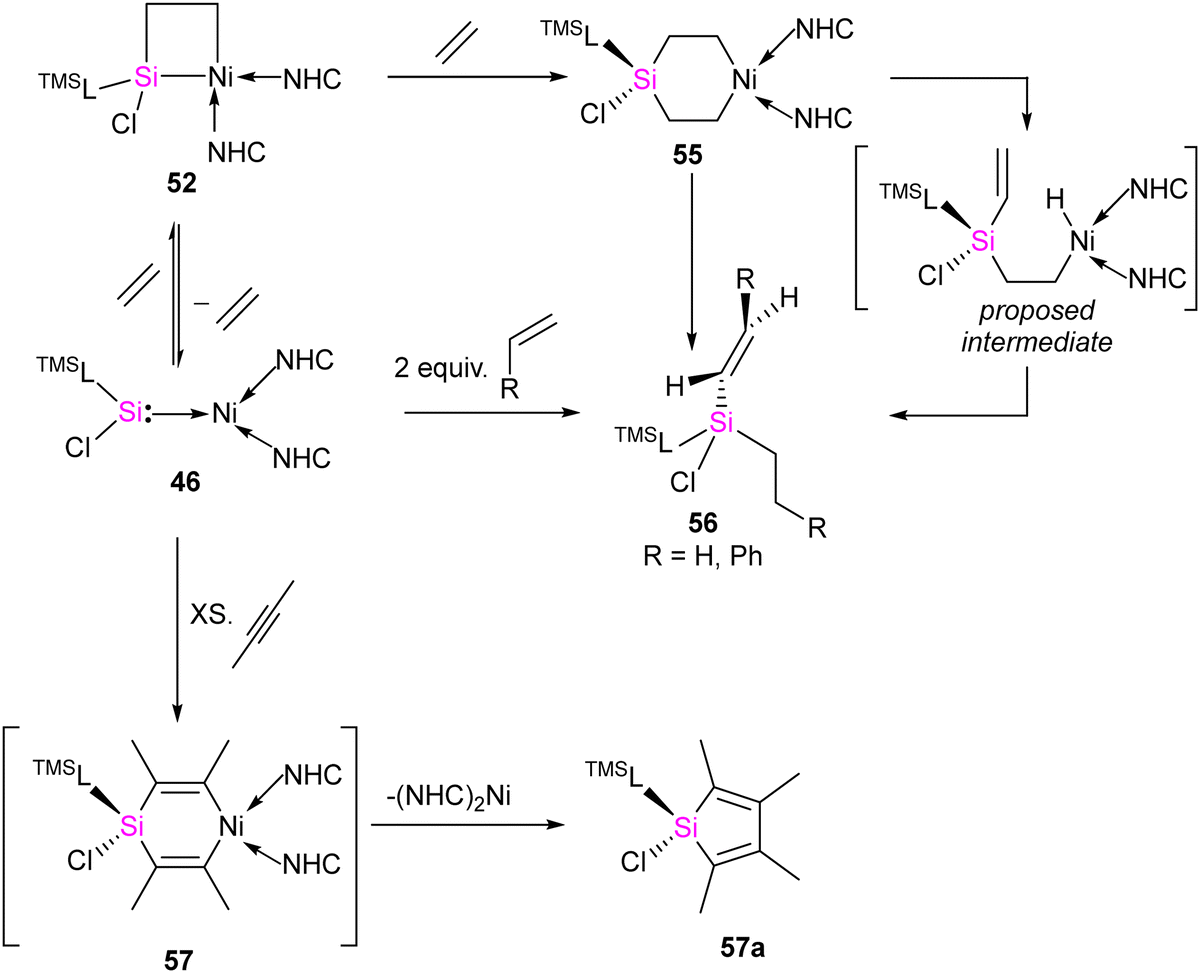 | ||
| Scheme 20 The reaction of 46 with alkenes. | ||
Previous examples of silylene–Ni complexes discussed above are based on monodentate silylene ligands. In 2021, Xi and co-workers synthesized a novel phosphine–silylene mixed donor ligand and studied its coordination chemistry with Ni(0).67 The reaction of [(PhC(NtBu)2SiCl)] with Li[(3,5-Me2-C6H3)NPiPr2] in THF afforded the desired mixed donor ligand 58 in good yield. Treatment of 58 with Ni(COD)2 in a 1:1 molar ratio resulted in a κ2-P, Si–Ni complex 59, where the ligand acts as a bidentate ligand (Scheme 21). Interestingly, the reaction of 59 with Ad–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P resulted in the formation of a unique 1,3-diphosphacyclobutadiene complex 60via Ni(0)-mediated selective head-to-tail cyclization of two phosphaalkynes (Scheme 21). The 29Si NMR spectrum of 58 (−58.85 ppm) is downfield shifted to those of Ni(0) complexes 59 (δ 11.34 ppm) and 60 (δ 17.44 ppm), respectively.
P resulted in the formation of a unique 1,3-diphosphacyclobutadiene complex 60via Ni(0)-mediated selective head-to-tail cyclization of two phosphaalkynes (Scheme 21). The 29Si NMR spectrum of 58 (−58.85 ppm) is downfield shifted to those of Ni(0) complexes 59 (δ 11.34 ppm) and 60 (δ 17.44 ppm), respectively.
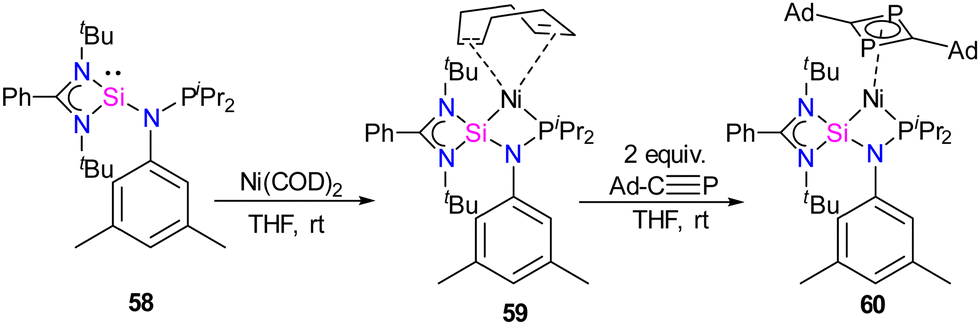 | ||
| Scheme 21 (a) Synthesis of phosphine functionalized silylene–Ni(0) complex 59. (b) The reactivity of 59 with Ad–CP. | ||
The molecular structures of 59 and 60 show that the Ni center adopts distorted tetrahedral geometry with cyclooctadiene/1,3-diphosphacyclobutadiene occupying one of the coordination sites (Fig. 13).
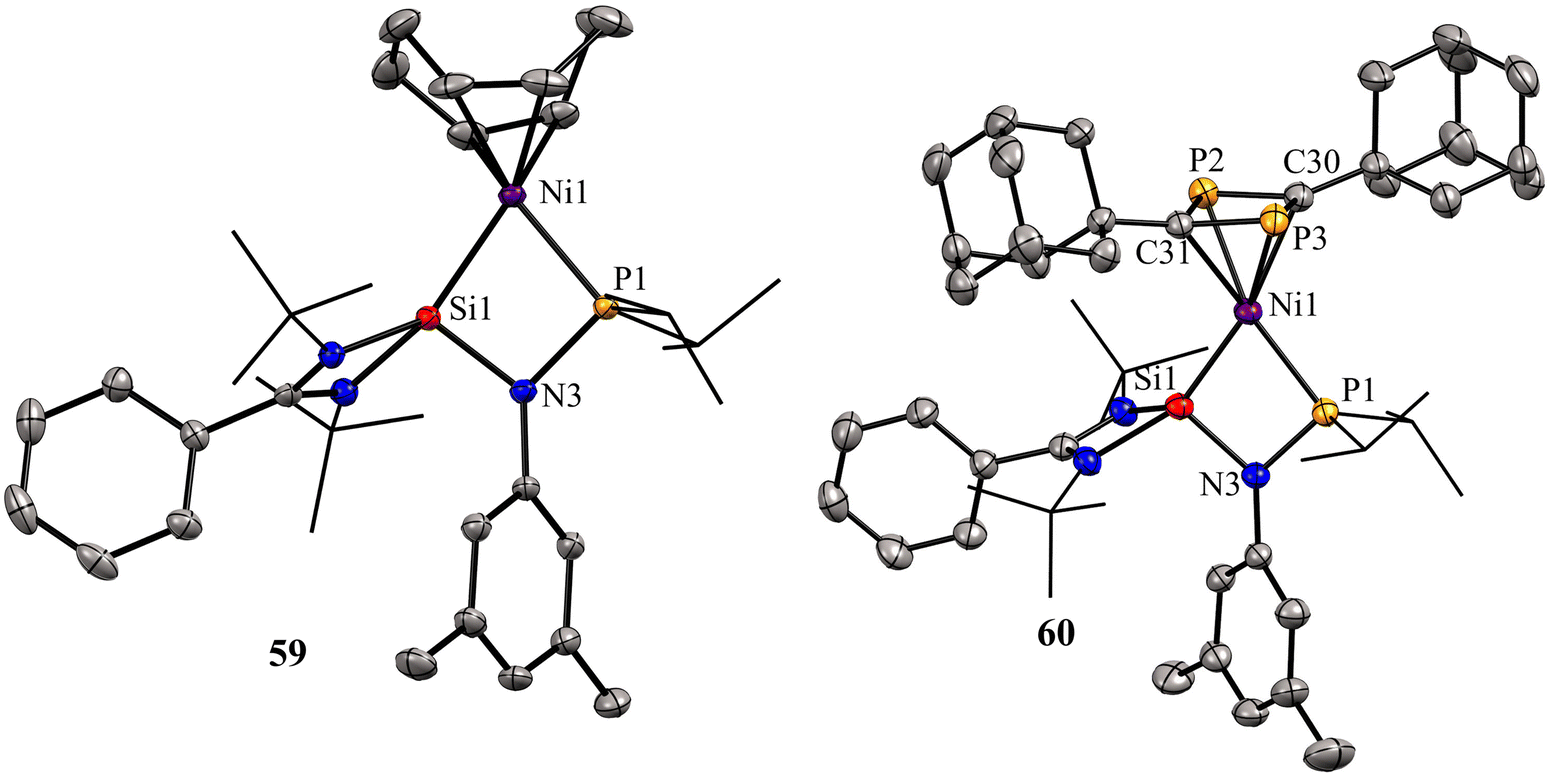 | ||
| Fig. 13 Molecular structures of 59 and 60. Reproduced from ref. 67 Copyright©2021, American Chemical Society. | ||
In 2022, Khan and co-workers designed and synthesized novel phosphine-silylene hybrid ligands 11 and 61 (Scheme 22) and their transition metal halide complexes.35 The ligands 11 and 61 were easily obtained by the reaction of [(PhC(NtBu)2SiCl)] with LiN(R)(C6H4)PPh2 (R = TMS, TBDMS). The 31P and 29Si NMR spectra of ligands displayed up-field shifted chemical resonances compared to those reported for similar hybrid silylenes, suggesting this might be due to the presence of strong electron donating TMS/TBDMS group on the N atoms in 11 and 61, respectively. They investigated the reactivity of these ligands with MX2 complexes (M = Fe(II), Co (II), and Ni(II)), and their electrochemical, optical, and magnetic properties were also explored. As shown in (Scheme 22), the reaction of 11 and 61 with NiX2·dme (X = Cl, Br), (dme = ethylene glycol dimethyl ether), afforded the desired phosphine–silylene–Ni(II) complexes 62 and 63.
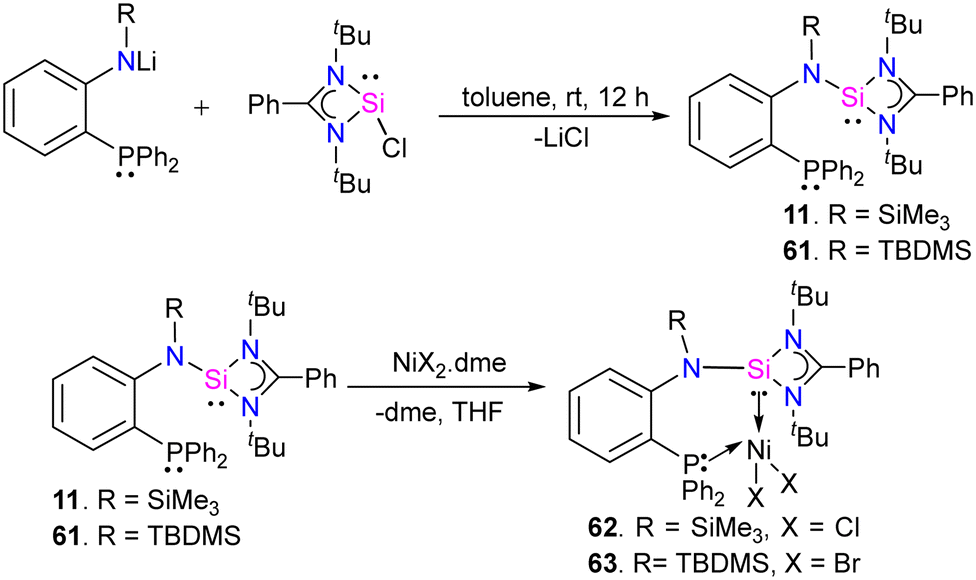 | ||
| Scheme 22 (a) Synthesis of 11 and 61. (b) The reaction of 11 and 61 with NiX2·dme. | ||
The 31P-NMR and 29Si-NMR spectroscopies of 62 and 63 demonstrated a downfield shift in comparison with corresponding phosphine–silylene ligands that is attributed to the decrease of electron density on P and Si centers after coordinating to Ni(II) center. Furthermore, the molecular structures of 62 and 63 showed a disordered square planar geometry around Ni(II) metal. Complexes 62 and 63 were thermally more stable up to 290 °C.
Cyclic voltammetry studies on 62 showed distinct redox peaks, suggesting an electroactive nature of 62. At the same time, the initial phosphine–silylene ligand did not demonstrate such a property. In the CV analysis of 62, two reversible redox events were identified with reduction peaks at Epc = −1522 mV and Epc = 1960 mV, corresponding to Ni(II) → Ni(I) and Ni(I) → Ni(0), respectively. The associated oxidation peaks were observed at Epa = −1950 mV and Epa = −1611 mV, corresponding to Ni(0) → Ni(I) and Ni(I) → Ni(II), respectively (Fig. 14).
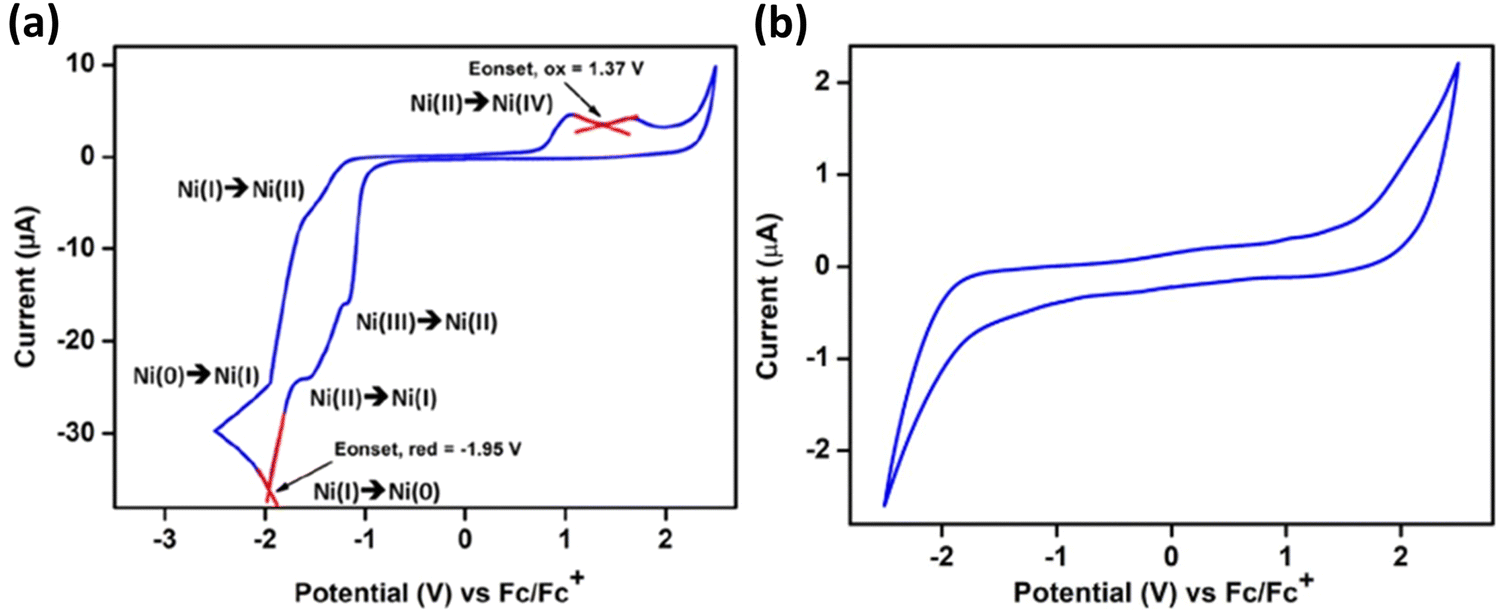 | ||
| Fig. 14 CV of complex 62 in CH2Cl2(a) and ligand 61 in THF (b), with 0.05 M t-butyl-ammonium-hexafluorophosphate. All potentials were referenced to the Fc/Fc+ couple. Scan rate = 50 mV s−1. Reproduced from ref. 35 Copyright©2022, American Chemical Society. | ||
A reduction peak at Epa = −1172 mV was also assigned to the Ni(III) → Ni(II) electron transfer event. The broad oxidation peak at an extreme positive potential indicates a Ni(II) → Ni(IV) two-electron oxidation with Epa values of 1054 mV and 1708 mV, respectively. Although the standard oxidation potential of Ni(II) to Ni(IV) is Epa = 1590 mV, a dual peak behaviour is observed when Ni(II) directly oxidizes to Ni(IV). Similar redox behaviour was noted for compound 63.
Studies on magnetic properties have disclosed that complexes 62 and 63 exhibit magnetization under an external magnetic field. A magnetic moment of zero is anticipated in a four-coordinate Ni(II) complex with a square planar geometry. Nevertheless, substantial distortion in this geometry can induce magnetization in the complex. As a result of such distortion, complexes 62 and 63 exhibit magnetic moments of 1.75 and 1.4 Bohr magnetons (BM), respectively, affirming their paramagnetic nature at room temperature and superparamagnetic behavior at low temperatures. This marks the first example of a silylene-supported nickel(II) complex showcasing superparamagnetic behavior. This phenomenon is linked to the distorted square planar geometry, which, in turn, is influenced by the structure of the ligand. The magnetic measurement studies suggest that the complexes show super magnetic character at low temperatures, suggesting that with future generations, silylene-based ligands could provide a unique opportunity to be utilized in material science for various applications.
Transition metals, including Ni, typically act as Lewis acids but can also function as Lewis bases, which is less common.68,69b Metallylene ligands from group-14 (R2E:) (E = C, Si, Ge, Sn, Pb), such as carbene and its heavier analogs, exhibit an ambiphilic character featuring a divalent center with a lone pair orbital (nσ) and a vacant orbital (pπ). This ambiphilic nature leads to two potential coordination modes through σ-electron donation: (i) from R2E to metal [R2E: → M],3b,19c resulting in classical complexes with a planar geometry around the E atom, classified as Fischer or Schrock-type complexes; or (ii) from metal to ER2 [M → ER2],69 giving rise to non-classical metallylene complexes characterized by a strongly pyramidalized E center. Base-stabilized metallylenes exhibit [R2E:→M] bond formation due to the significantly lower Lewis acidity of the E atom compared to their base-free counterparts. In contrast, base-free metallylenes are more prone to π-backdonation (M→ER2), resulting in non-classical metallylene complexes with ME double bond character. Descending a group in the periodic table influences the nucleophilicity of divalent atoms (E), resulting in a decrease due to an increase in the s-character of the lone pair. Simultaneously, the unoccupied pπ orbital becomes more Lewis acidic. Consequently, heavier divalent species (E = Ge, Sn, Pb) tend to form M → ER2 complexes stronger. The only known compounds of this type are based on germylene, stannylene, and plumbylene complexes.69 A non-classical novel metallylene complex, stabilized by σ-donating Ni(0) ligand coordination, was introduced by Kato and co-workers in 2022 (Scheme 23).70 Complex 65 is an unusual 16 VE-Ni(0)–silylene complex, displaying distinct characteristics of non-classical metallylene complexes. It features a strongly pyramidalized and nucleophilic divalent silicon center, setting it apart from conventional coordination structures.
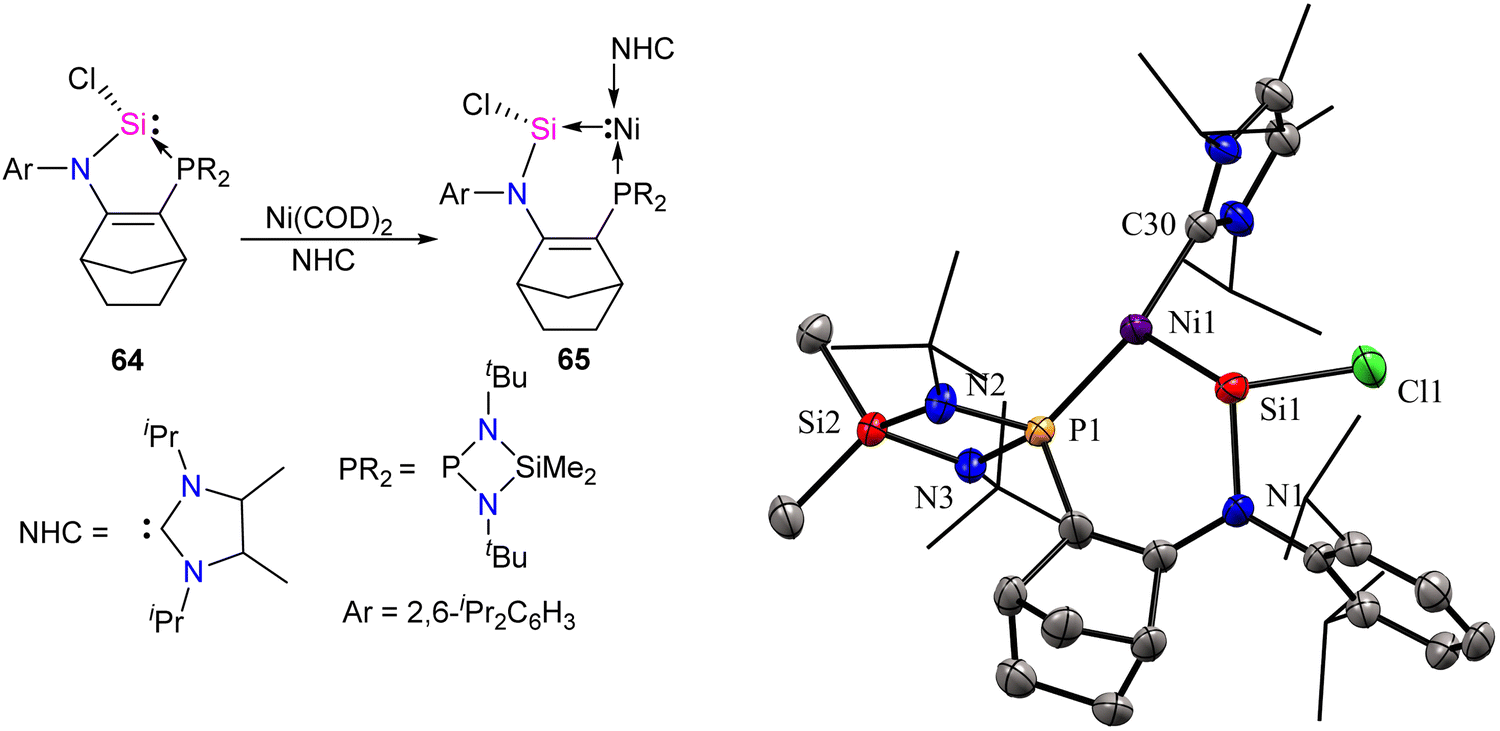 | ||
| Scheme 23 Synthesis of Ni-stabilized silylene 65 and its molecular structure. Reproduced from ref. 70 Copyright©2022 Wiley-VCH GmbH. | ||
The molecular structure of 65 reveals an elongated Si–Ni bond (2.178 Å) compared to other Ni(0)–silylene complexes (2.075–2.133 Å).70 This value is within the range of Ni–Si single bonds.63 These structural data of 65 agree with a non-classical complex (Ni → silylene) with a lone pair on the Si atom and a reduced Si–Ni multiple bonding character. Further, the authors also explored the reactivity studies of 65 with various small molecules and organic spacers (Scheme 24).
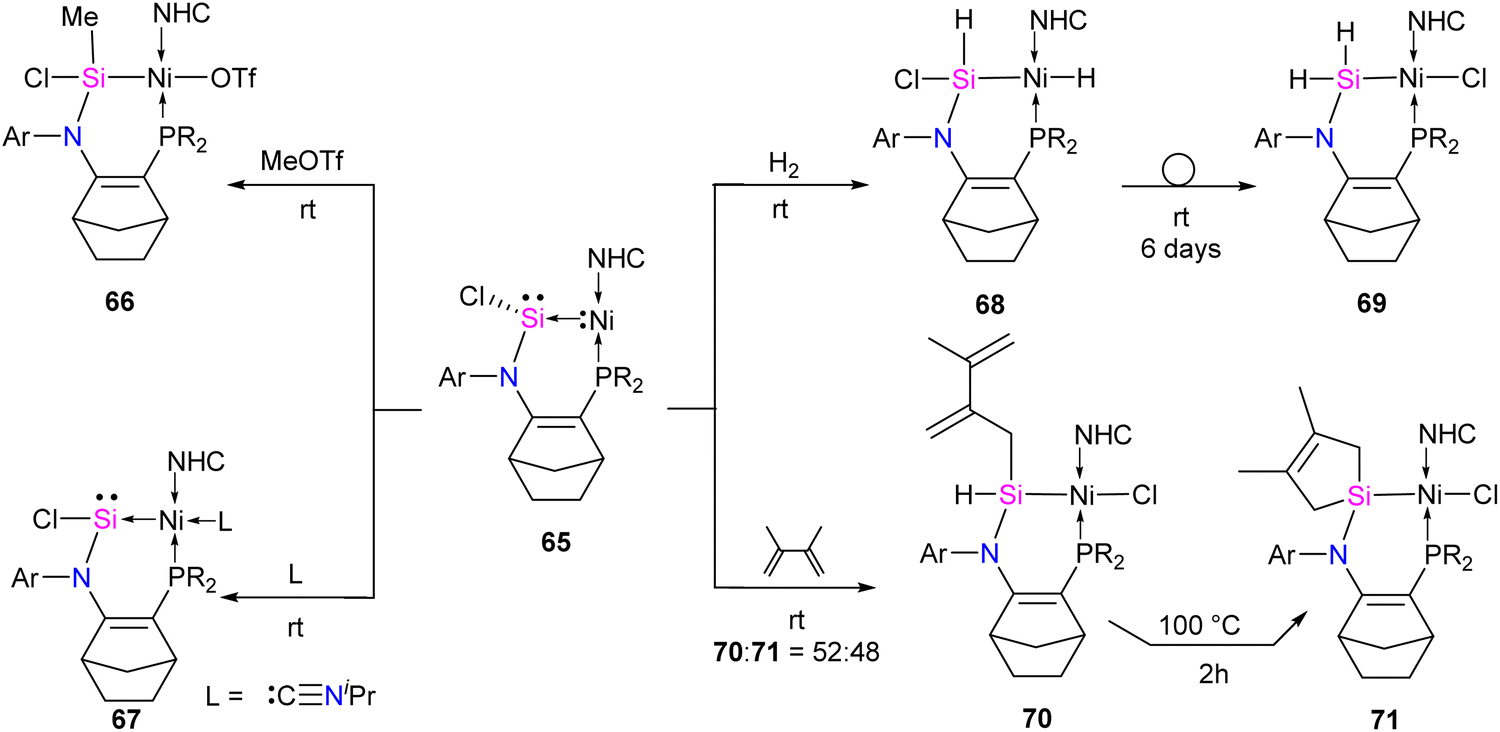 | ||
| Scheme 24 Reactivity studies of Ni-stabilized chlorosilylene complex 65. | ||
The reaction of 65 with MeOTf led to the formation of Si–methylated Ni(II) complex 66 (Scheme 24). This highlights the nucleophilic character present at the silicon center, which is, in contrast, the electrophilic nature of the Si center in classical silylene–TM complexes. Furthermore, when a Lewis base, such as isopropyl isocyanide, coordinates with the metal center, it forms a tetra-coordinate Ni(0) complex 67. The distinctive characteristics of complex 67 include a Si(II) center that is less pyramidalized (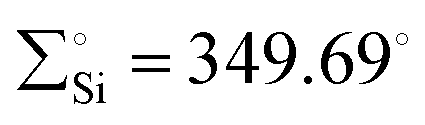 ) and a shorter Si–Ni bond [2.1108(5) Å], in contrast to 65 [
) and a shorter Si–Ni bond [2.1108(5) Å], in contrast to 65 [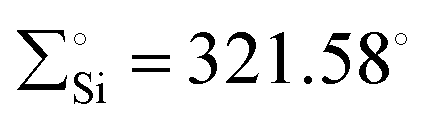 , Si–Ni: 2.1780(7) Å]. This implies an increased Si–Ni π-back donation in 67, potentially attributed to geometrical modifications at the Ni(0) center (transition from T-shape to distorted tetrahedral). Silylene complex 65 reacts with H2 at room temperature, forming a formal 1,2-dihydrogen adduct 68. Over time, this adduct slowly undergoes a gradual isomerization, generating the corresponding isomer 69. This isomerization process involves the exchange of substituents between H and Cl on the Si and Ni atoms, as depicted in Scheme 24.
, Si–Ni: 2.1780(7) Å]. This implies an increased Si–Ni π-back donation in 67, potentially attributed to geometrical modifications at the Ni(0) center (transition from T-shape to distorted tetrahedral). Silylene complex 65 reacts with H2 at room temperature, forming a formal 1,2-dihydrogen adduct 68. Over time, this adduct slowly undergoes a gradual isomerization, generating the corresponding isomer 69. This isomerization process involves the exchange of substituents between H and Cl on the Si and Ni atoms, as depicted in Scheme 24.
The silylene–Ni complex 65 reacts rapidly with 2,3-dimethyl-1,3-butadiene at room temperature, producing a mixture containing two Si(IV)Ni(II) complexes, 70 and 71, in a 1:1 ratio. These complexes are formally generated through a C–H insertion or a [4+1] cycloaddition at the Si center. This is followed by a 1,2-migration of the chlorine atom to the Ni center, as outlined in Scheme 24. Complex 70 undergoes isomerization at 100 °C over 2 hours to produce 71.
The reaction of 65 with PhLi was conducted to investigate the substituent effect on the reactivity/stability of the Ni → Si complexes. This reaction produced the corresponding phenyl-substituted silylene complex 72, as outlined in Scheme 25. Complex 72 exhibits instability at temperatures above −30 °C. It undergoes C–H activation of the NHC motive across the Si–Ni fragment, forming the silyl hydride Si(IV)Ni(II) complex 73. Upon warming the reaction mixture to ambient conditions, complex 74 undergoes further isomerization through the exchange of ligands (H and CH2) on the Si and Ni centers, resulting in the formation of a stable pincer-type nickel(II) hydride complex 75 (Scheme 25).
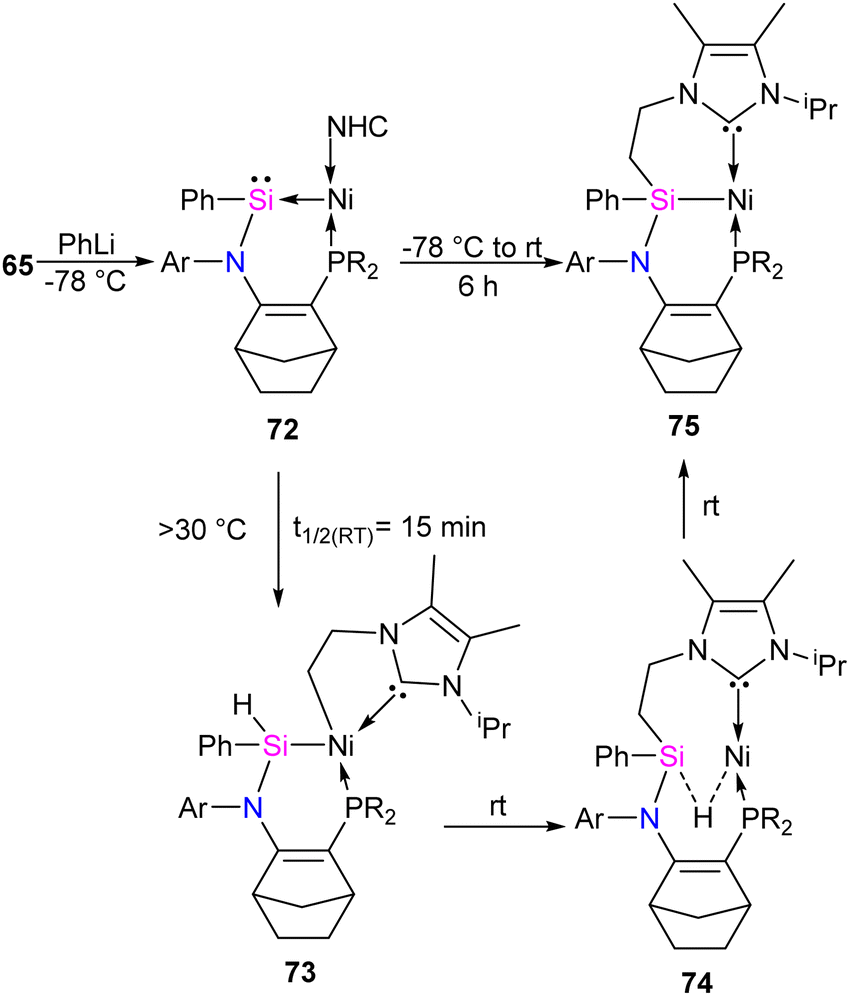 | ||
| Scheme 25 Synthesis of phenyl-substituted silylene 72 and its isomerization. | ||
Compound featuring Pd, Pt–Si (silylenes) bond
Palladium (Pd) and platinum (Pt) complexes with silylene ligands can be synthesized as either mononuclear or multinuclear compounds bridged by silylene motifs. These complexes are utilized in various applications, including cross-coupling reactions and small molecule activations, owing to their distinctive electronic and steric characteristics (Fig. 15).71 This class of compounds has attracted considerable attention due to its structural and catalytical properties, especially in producing organosilicon compounds and polysilane(s).72 Different approaches for preparing Pd, and Pt–silylene complexes have been introduced to date, anion abstraction, photolysis, trapping method, dehydrogenative condensation approach, and the direct reaction of isolabel silylene with metal complexes.71f,73 The first example of platinum silylene complex [trans-(Cy3P)2(H)PtSi(SEt)2][BPh4], was synthesized by Tilley and Rheingold in 1993.71d This cationic Fischer-type complex was prepared via anion abstraction from a platinum silyl precursor.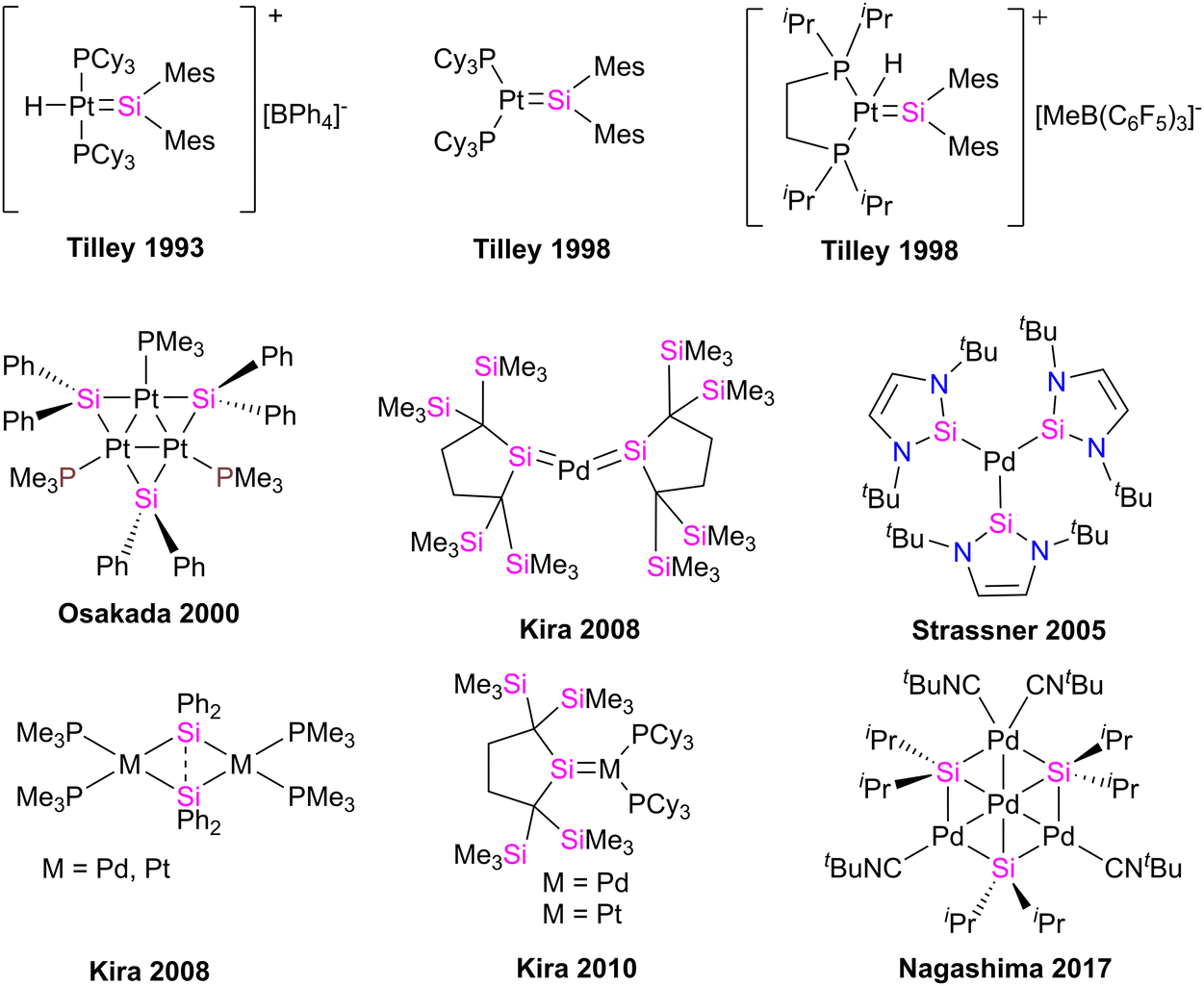 | ||
| Fig. 15 Selected examples of Pd, Pt complexes of silylenes. | ||
In a recent work by Hinz, the coordination behaviour of the carbazolyl stabilized bromosilylene 76 toward Pt metal and their reactivity with ethylene has been investigated.74 As shown in Scheme 26, the product formation depends on the platinum source used in the reaction. The reaction of 76 with [(η2-C2H4)Pt(PPh3)2] resulted in the formation of a four-membered platinasilacyclobutane 77 with a tetra-coordinated silicon atom. The reaction proceed via the formation of platinum–silylene R(Br)SiPt(PPh3)2 complex which further undergoes a [2+2] cycloaddition with the ethylene released during the reaction to afford 77. Furthermore, it was demonstrated that 77 could be expanded to a cyclohexane-like structure 78 by the insertion of another ethylene into the Pt–Si bond. On the other hand, when the bromosilylene 76 reacted with Pt(PCy3)2, the formation of R(Br)SiPt(PCy3)2 (79) was observed. The reaction of 79 with ethylene gas led to the formation of a six-membered platinasilacycle R(Br)Si(C2H4)2Pt(PCy3)2 (80). The compound (79) is not stable in the solution and at room temperature, and within one day, it decomposed and resulted in the formation of a free Pt(PCy3)2 and a silicon-containing decomposition product (79-I) (Scheme 27). The proposed mechanism for the decomposition of the product suggests that it proceeds through the π-coordination of one of the flanking arene moieties to the low-coordinated silicon center. Subsequently, activation of the C–H bond at the silicon center occurs, attributed to the increased acidity of the silicon atom upon coordination with the metal fragment. As a result, the Pt complex can dissociate readily, generating a Si(IV) compound.
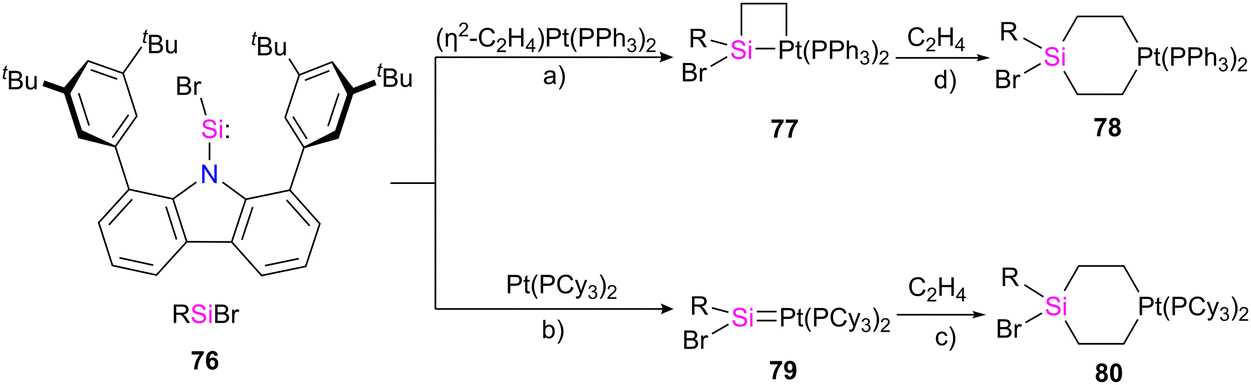 | ||
| Scheme 26 Synthesis of compounds 77–80 starting from RSiBr 76. Reaction condition: (a) toluene, RT, 12 h; (b) toluene, RT, sonication 15 min; (c) toluene, 1 atm C2H4, RT, 12 h; (d) C6D6, 1 atm C2H4, 80 °C, 72 h. | ||
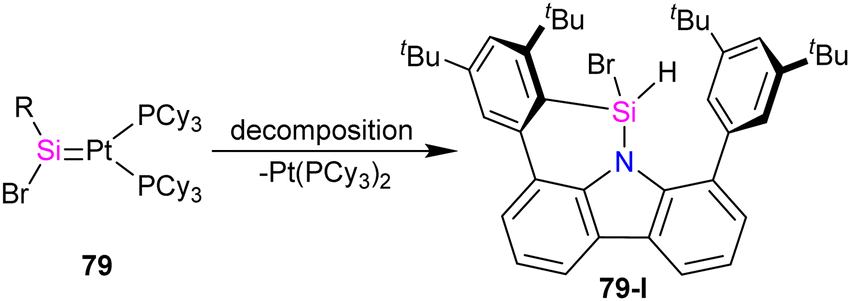 | ||
| Scheme 27 Decomposition of 79 to 79-I. | ||
In another study conducted by Osakada and collaborators, di- and trinuclear complexes featuring Pd(0) and Pt(0) with bridging silylene ligands were synthesized, and their reactivity towards alkynes was systematically examined.75 Prior knowledge indicated that the reaction of (aminosilyl)boronic esters with Pt(0) and Pd(0) complexes leads to the formation of mono- and dinuclear complexes containing bridging silylene ligands. Building on this understanding, the researchers employed various ratios of (aminosilyl) boronic ester 81 and Pt(0), Pd(0) precursors to create a diverse set of di- and trinuclear metal–silylene complexes, as illustrated in Scheme 28. This strategic variation in reactant ratios allowed for the exploration of different synthetic pathways and the generation of a range of metal–silylene complexes, contributing to a deeper understanding of their reactivity and potential applications.
 | ||
| Scheme 28 Preparation of di- and trinuclear Pd(0)/Pt(0) complexes with bridging silylene ligands. | ||
To get more insight into the chemistry of the synthesized dinuclear Pt(0) and Pd(0) compounds, the reactivity of the silylation of alkynes was investigated in this work. Based on this study, the stoichiometric reaction of 82-Pt2 with terminal alkynes such as HCCtBu and HCCSiMe3 at room temperature resulted in the formation of diplatinum complexes with hydride and bridging alkynyl ligands (84a, 84b) (Scheme 29). The reaction involves C(sp)–H bond activation of the terminal alkyne by the Pt center and π-coordination of the resulting alkynyl ligand to another. Further reaction of 84b with the mentioned alkynes resulted in the alkyne insertion into the Pt–Si bond and coupling the resulting alkenyl carbon bonded to Pt and the hydride ligand to form 85a and 85b. Therefore, this reaction can be regarded as hydrosilylation of the alkyne by the hydride and bridging silyl ligand (Scheme 29). This hydrosilated alkyne was easily separated from platinum fragment by adding 1,2-bis(diphenylphosphino)ethane (dppe) to diplatinum complex 85b resulted in the reductive elimination of Me3SiCCSi(Ph)2C(SiMe3)CH2 (86b, 68%), which was accompanied by formation of [Pt(dppe)2] (Scheme 29).
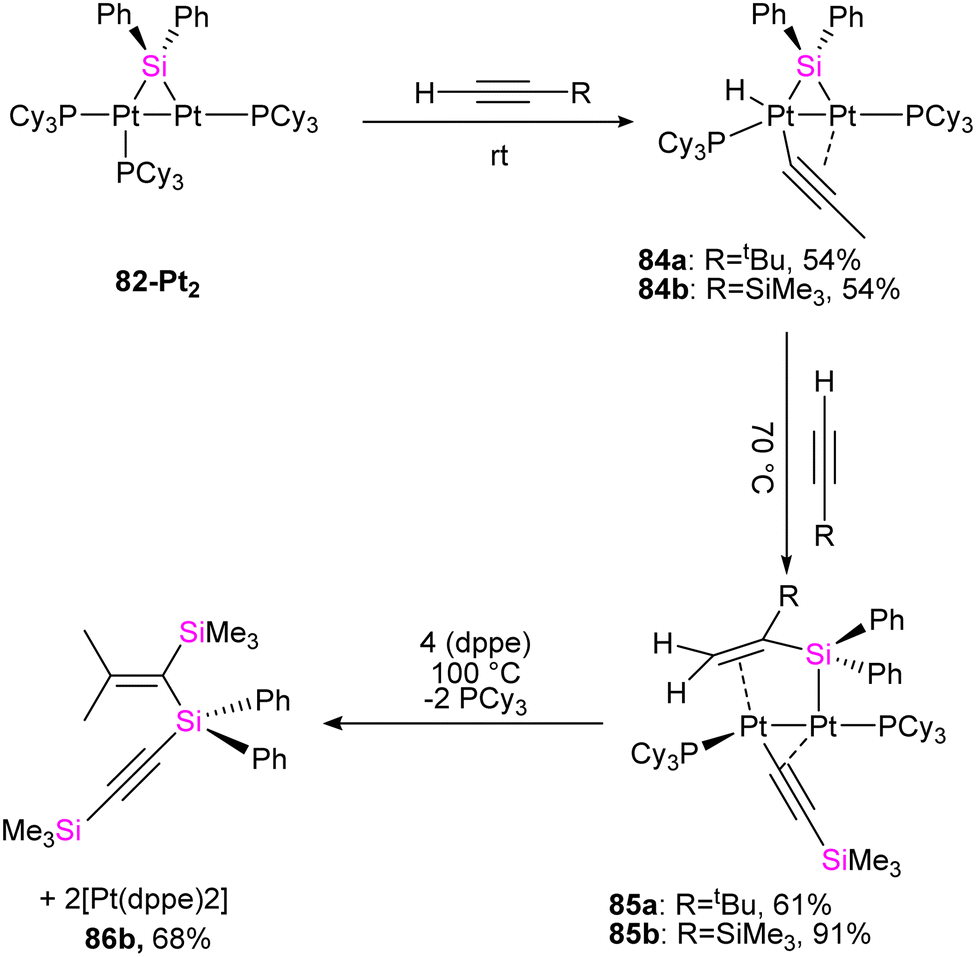 | ||
| Scheme 29 The stoichiometric reaction of 82-Pt2 with terminal alkynes. | ||
The reactivity of 82-Pt2 with internal alkynes was also studied in this work. The silylation of the alkyne group, followed by the formation of their alkyne-coordinated Pt(0) complexes 87 was observed when the alkyne precursor does not have any reactive group near to the triple bond (Scheme 30(a)). In contrast, in the case of the reaction of a 1:3 molar ratio of 82-Pt2 with dimethyl acetylenedicarboxylate (DMAD), which has carboxylate group as a reactive group in its formula, a diplatinum complex with a silaplatinacyclohexadiene structure 88 was produced (Scheme 30(b)). This group also used the dipalladium complex 82-Pd2 to demonstrate the catalytic reaction of alkynes with dipalladium complex 82-Pd2 and compared the result with mononuclear Pd–silylene complexes.
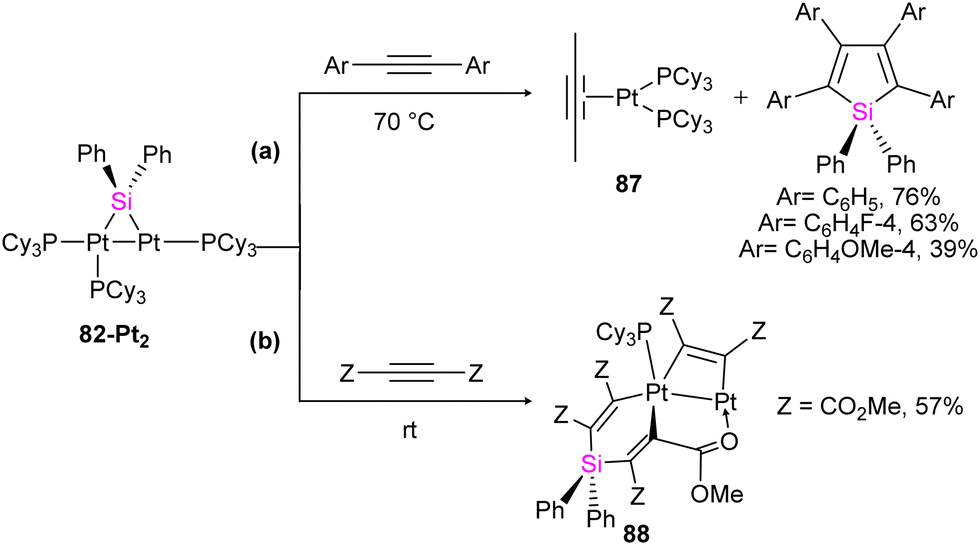 | ||
| Scheme 30 The reaction of 82-Pt2 with internal alkynes. | ||
The catalytic ability of dipalladium complex 82-Pd2 in the silylation of alkynes with (amino silyl) boronic ester 81 was also examined in this work. And it was shown that 82-Pd2 could catalyze 1:2 cyclo coupling of Et2NSiPh2B(pin) with monosubstituted acetylenes to form 2,4-disubstituted silole (89) as the major product as well as by-products including 3,4-disubstituted silole (90) and alkynyl(alkenyl)silane (91) (Scheme 31). The yields of the products, depended on the amount of solvent, reaction temperature, and addition of the PCy3 ligands.
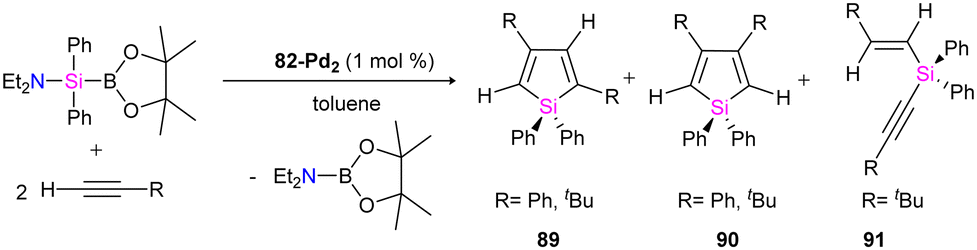 | ||
| Scheme 31 The catalytic reaction of 82-Pd2 in the silylation of alkynes. | ||
The possible mechanism for forming the product and the by-products is demonstrated in Schemes 32 and 33, respectively. As shown in Scheme 32, the reaction is carried on by the coordination of the alkyne molecule to the Pd center in the first step (A), followed by the insertion of this alkyne into the Pd–Si bond of the bridging silylene ligand. Further insertion of another alkyne molecule to the remaining Pd–Si bond gives a dipalladasilacyclopentene intermediate (B) and its regioisomer (C). The resulting siladipalladacycloheptadienes (B and C) undergo a 1,2-reductive elimination of the silole to form the products (89 and 90).
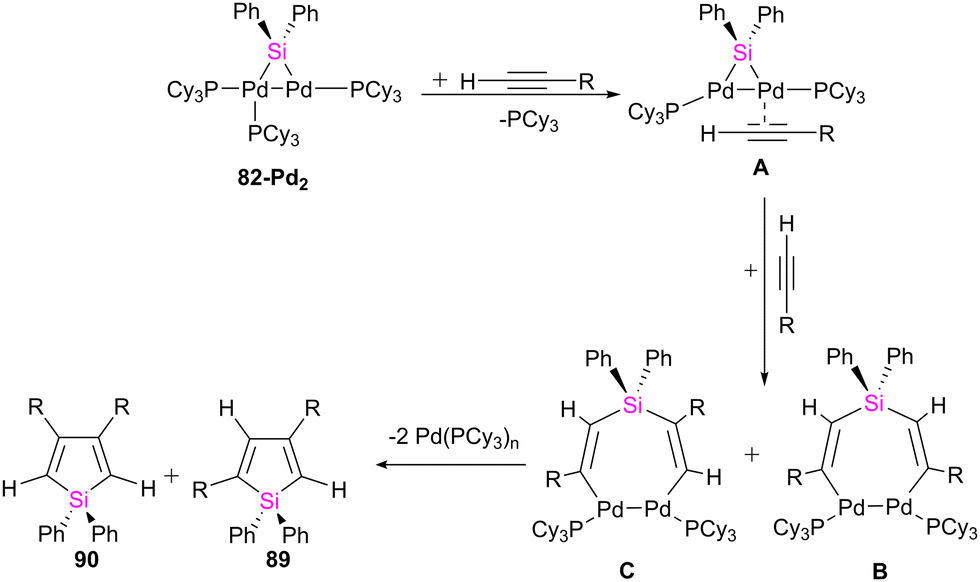 | ||
| Scheme 32 Possible pathway for the formation of siloles from 82-Pd2 and terminal alkynes. | ||
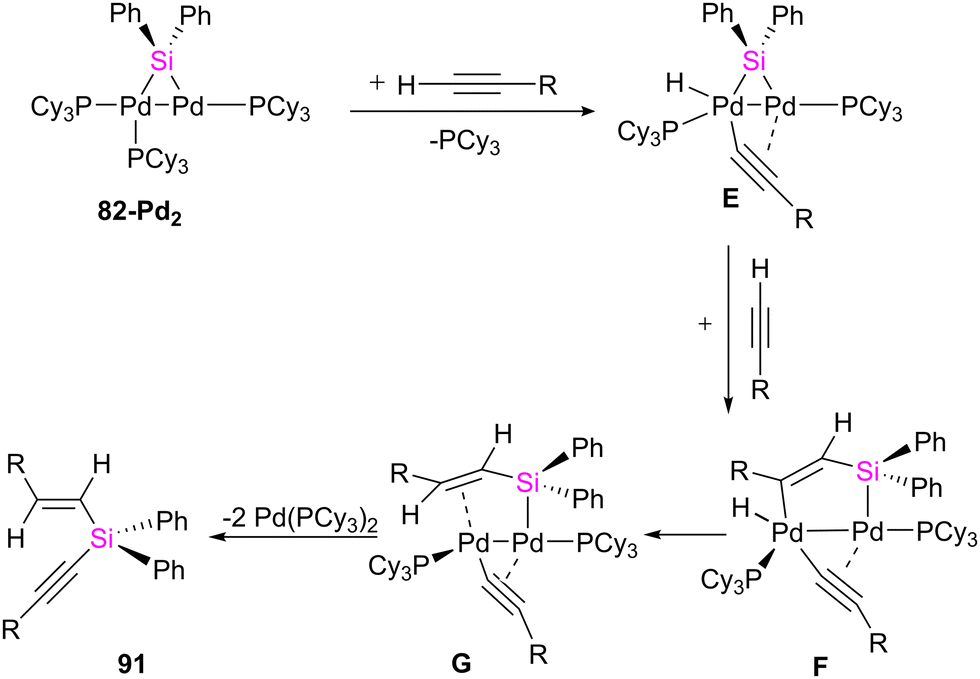 | ||
| Scheme 33 Possible pathway for the formation of acyclic silane 91 from 82-Pd2 and terminal alkynes. | ||
Mono and dinuclear Pt/Pd complexes of silylenes and their structural and catalytical properties have been extensively studied, whereas trinuclear and multinuclear complexes incorporating silylene ligands are rare.17d Given this, Nakata and co-workers explored the coordination chemistry of chlorosilylene 92 with Pd(PPh3)4 and [PdMe2(tmeda)] (tmeda = N,N,N′,N′-tetramethylethylenediamine) as the metal precursors.76 The reaction of 92 with [Pd(PPh3)4] yielded a homoleptic tris(silylene)palladium(0) complex 93 through a ligand-exchange process (Scheme 34(a)). Whereas the reaction of 92 with [PdMe2(tmeda)] resulted in an unprecedented tetranuclear Pd4Si3 cluster featuring palladium atoms in different oxidation states (94) (Scheme 34(b)). The 29Si NMR spectra of 93 and 94 showed a doublet resonance centered at 75.8 and 184.2 ppm (JSiP = 9 Hz), downfield shifted compared to 92 (59.8 ppm). The molecular structure of 92 revealed a trigonal planar arrangement of silylene ligands around the Pd center. In contrast, those of 93 showed that the central six-membered (Pd and Si atoms) ring has a bowl shape (Fig. 16). Further, the X-ray photoelectron spectroscopy study suggested that the Pd centers in 94 are in different oxidation states (Pd(II) and Pd(0)). DFT calculations revealed that the three silicon atoms in cluster 94 serve as Lewis-base-stabilized silylene ligands, coordinating in a μ3-manner with both the outer and central palladium atoms.
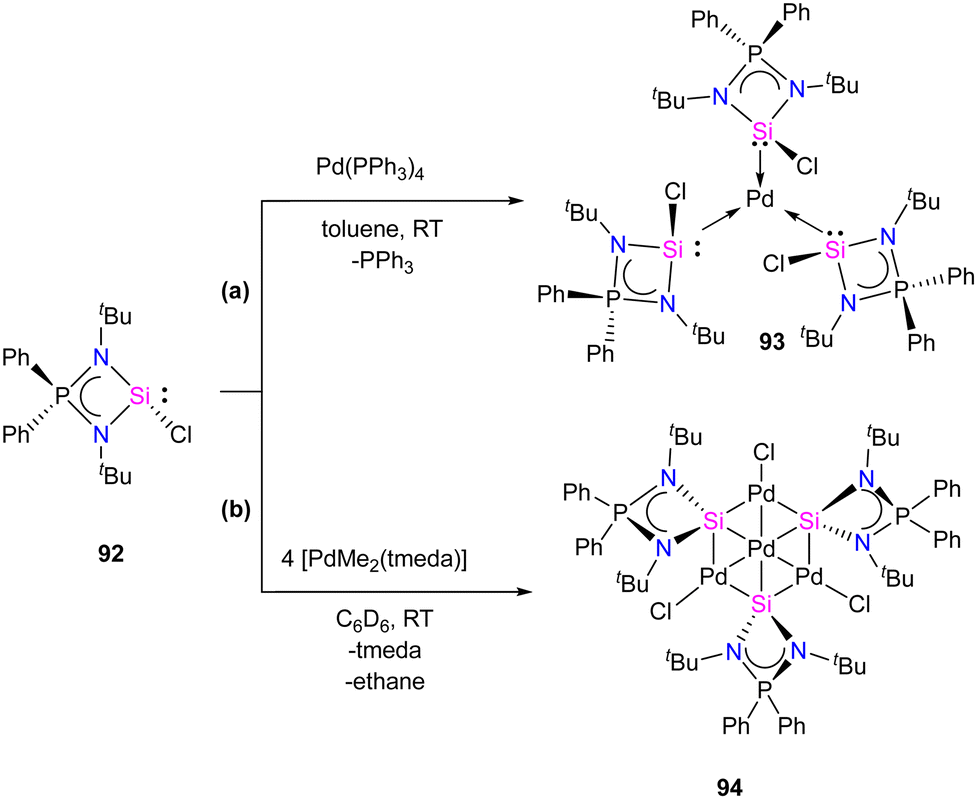 | ||
| Scheme 34 Synthesis of monometallic and tetrametallic Pd complexes 93 and 94 through the reaction with chlorosilylene ligand 92. | ||
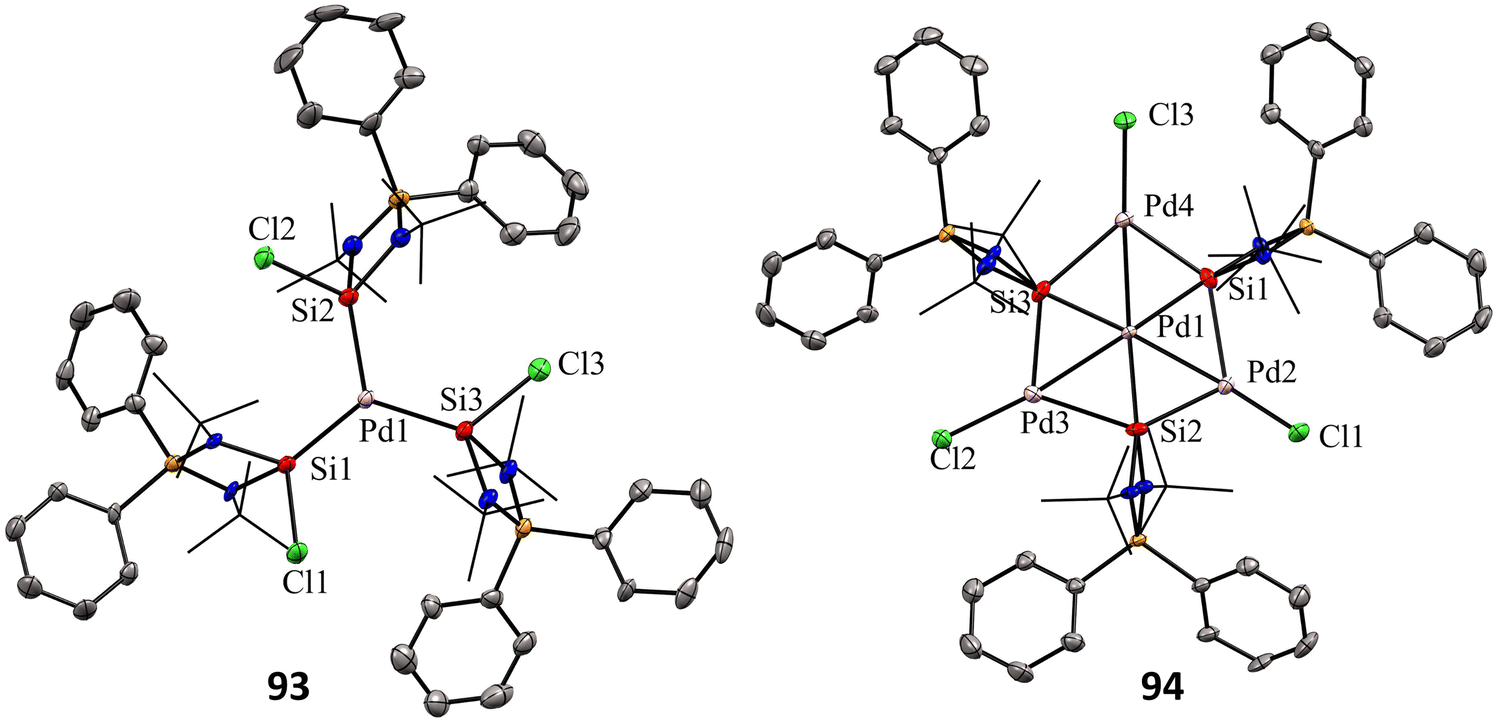 | ||
| Fig. 16 Molecular structures of 93 and 94. Reproduced from ref. 76. | ||
Coinage metal complexes
Silylene–coinage metal complexes are still in their early stages of development compared to their lighter counterparts. Given the promising results with NHCs, there is a pressing need to thoroughly investigate the chemistry of silylenes with coinage metals, as their unique electron donor and acceptor properties hold significant potential for diverse applications in the future.17a,77 Jutzi and co-workers were the first to isolate the first silylene coinage metal (Au) complex in 1990.78 Following this, Lappert and co-workers synthesized a Cu complex.79 Several silylene coinage metal complexes have been synthesized following these breakthroughs, and many of them have been utilized in various catalytic transformations (Fig. 17).17a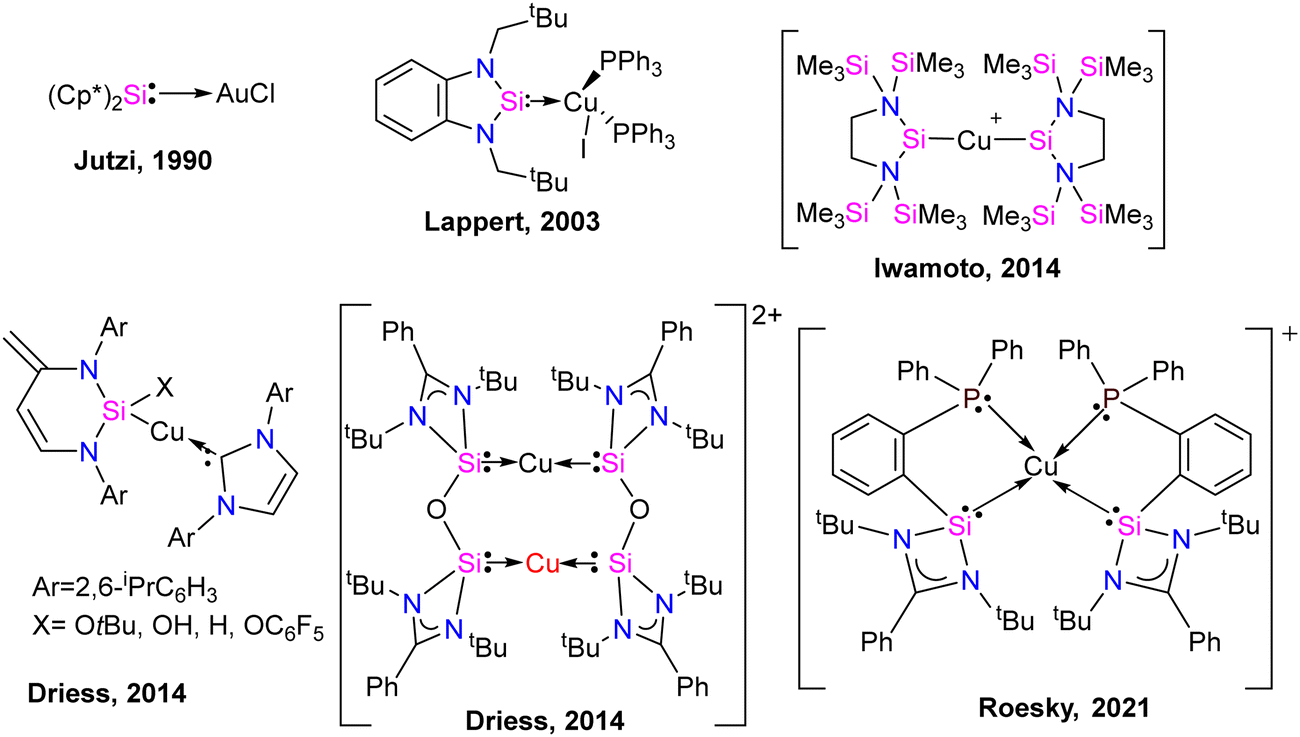 | ||
| Fig. 17 Selected examples of coinage metal complexes of silylenes. | ||
Valence tautomerism, or electromerism, is a well-known phenomenon in transition metal chemistry where electrons are redistributed between the metal and ligand without altering the structural motif.80 Valence tautomerism can be induced through external stimuli such as pressure, temperature, magnetic fields, or exposure to visible light or weak X-rays. However, such a phenomenon has only been recently reported in low-valent main-group chemistry.81 Driess and co-workers demonstrated that a redox non-innocent (bis)silylene-substituted ortho-carborane ligand stabilizes a zero-valent silicon species A and exhibits redox-induced electromerism. When compound A was subjected to one-electron reduction using KC8, the Si(0) center underwent formal oxidation to Si(I), while the ortho-carborane ligand backbone underwent two-electron reduction (Scheme 35(a)).82 Lately, phase-dependent electromerism in silylone C has been described by Iwamoto and co-workers (Scheme 35(b)).83
 | ||
| Scheme 35 Valence tautomerism in low-valent silicon chemistry. | ||
Very recently, P. Roesky and co-workers reported the first example of Lewis acid/base-induced reversible electromerism in low-valent silicon chemistry.84 A mixed-valent silaiminyl-silylene ligand [LSi-Si(NDipp)L] (L = PhC(NtBu)2) (96), was synthesized starting from LSiCl and DippNHLi (Dipp = 2,6-diisopropylphenyl) in toluene as a yellow solid in good yield (Scheme 36(a)). The 29Si{1H} NMR spectrum showed a resonance at δ = −61.7 ppm for (SiN) and at δ = 31.8 ppm for (silylene) silicon centers. This corroborates well with the oxidation states of Si centers in compound 95 as +I and +III, respectively. Interestingly, compound 95, on treatment with Lewis acidic copper salts, Cu(I)X (X = Mesityl, Cl, Br, I), resulted in the redistribution of oxidation states from +I and +III to +II for both the silicon atoms, leading to the formation of [{LSi(NDipp)Si(L)}CuX] (Scheme 36(b)). A singlet resonance in the 29Si NMR spectra (−9.7 to −5.9 ppm) and the molecular structure confirmed the bis-silylene coordinated copper complex formation.
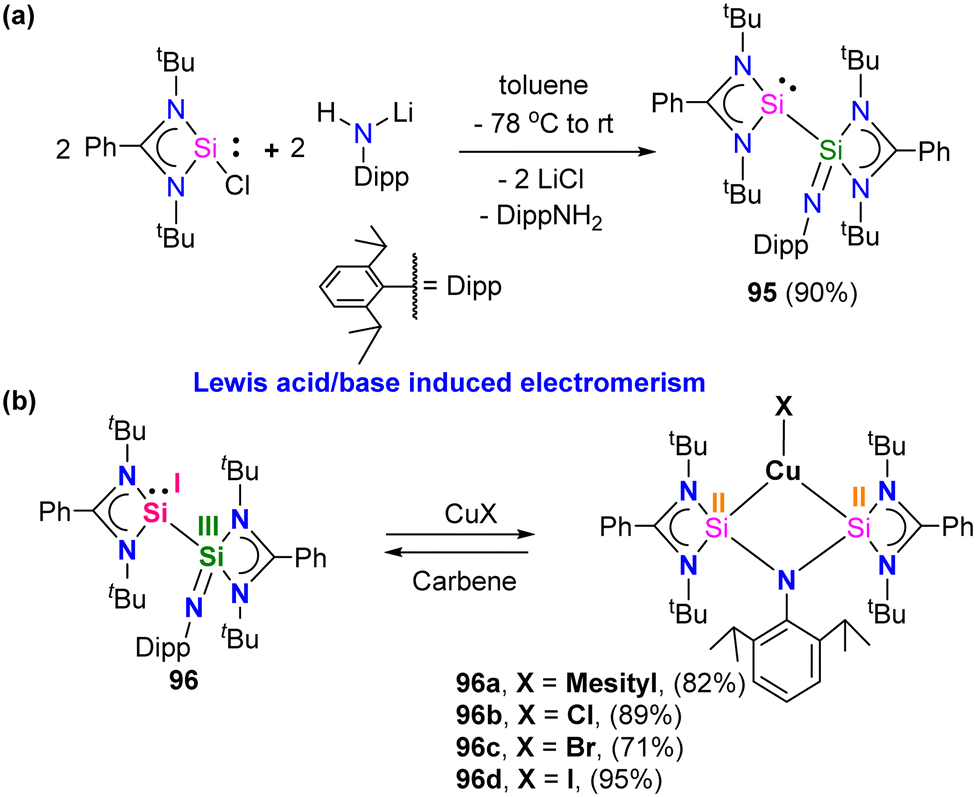 | ||
| Scheme 36 Synthesis of 95 and its copper(I) complexes. | ||
The authors hypothesized that the reaction proceeds via copper-coordination induced electromerisation of iminosilylsilylene 95. The electromerisation starts with the coordination of the lone pair on one of the Si atoms to the Cu center. Subsequently, the two-coordinate Cu center induces the redistribution of electrons, forming an additional silylene group, which coordinates with the Cu center to afford 96. Further, the authors also checked the reversibility of this process on an NMR scale reaction. When treated with strong Lewis basic free NHCs, the copper complexes resulted in the regeneration of iminosilylsilylene 95. This highlights that the stimuli-responsive nature of silaiminyl-silylene conversion might be useful for metal–ligand cooperation for bond-making and breaking processes during catalytic cycles. Recent years have seen a significant increase in interest in Au(I) complexes of phosphines and N-heterocyclic carbenes (NHCs) because of their significance in photophysical, biological application, and in catalysis.85 Au(I) complexes typically exhibit a preference for a linear geometry, which involves intra- or intermolecular aurophilic interactions. The coordination chemistry of Au(I) complexes has been greatly diversified by the efficient intra- and intermolecular aurophilic interactions, resulting in supramolecular structures and molecular aggregations in both the solid and solution phases.86 To optimize these interactions, it is most effective to utilize short-bite bidentate ligands, as they facilitate the close contact of metal ions. However, unlike phosphines and NHCs, the Au(I) complexes of silylenes have scarcely been studied. In this context, Nazish et al. utilized short-bite bidentate ligands having both phosphine and silylene as donors to synthesize dinuclear Au(I) complex with an Au⋯Au aurophilic interaction of 2.9987(7) Å.87 The reaction of phosphino–silylene 97 with two equivalents of AuCl(SMe2) in dichloromethane afforded the desired dinuclear Au(I) complex 98 in good yield (Scheme 37).
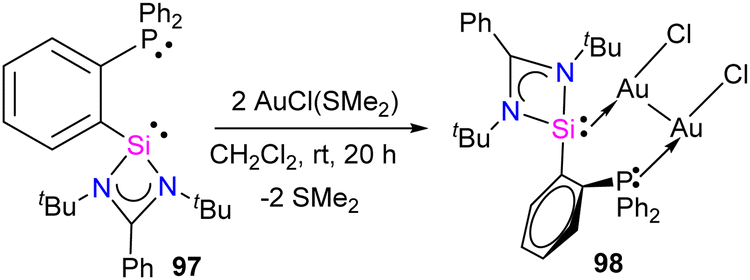 | ||
| Scheme 37 Synthesis of 98. | ||
The 31P NMR spectrum of compound 98 exhibited a singlet resonance at 25.57 ppm, which is downfield shifted compared to compound 97 (−11.18 ppm). This shift indicates the coordination of the phosphorus atom to gold (P → Au). The 29Si NMR spectrum of compound 98 showed a doublet centered at 1.97 ppm (JSi–P = 45.5 Hz) that is upfield shifted compared to compound 97 (18.52 ppm), indicating coordination of a Si(II) atom to an Au center. This upfield shift in the 29Si NMR spectrum may be attributed to back-donation from the Au(I) center to the Si(II) atom, resulting in an increase in electron density at the Si(II) atom. The molecular structure of 98 showed Au⋯Au aurophilic interaction of 2.9987(7) Å, resulting in a six-membered C–Si–Au–Au–P–C ring. Quantum chemical calculations were performed to gain insight into the bonding nature of complex 98. QTAIM analysis showed a bond-path and bond-critical point for the Au⋯Au aurophilic interaction. The EDA-NOCV calculation indicates that the most significant orbital interaction, ΔEorb1 (−58.0 kcal mol−1), arises from the σ-donation of Si(II) lone pair of electrons to the Au-atom of AuCl, resulting in the formation of a Si → Au dative bond. The second most significant interaction, ΔEorb2 (−44.2 kcal mol−1), arises from the σ-donation of the P(III) lone pair of electrons to the Au-atom of the other AuCl moiety, resulting in the formation of a P → Au dative bond. Hence, the silylene and phosphane components exhibit a synergistic effect in their coordination with Au(I) in complex 98. The third contribution Δρ3 primarily originates from the π-backdonation of the AuCl moiety to the Si(II) and P(III) sites, together with Au⋯Au orbital–orbital interactions, which cannot be exactly distinguished.
The Group-10 metal complexes stabilized by silylenes have gained recent interest. However, they are rarely explored in homogeneous catalysis.88 These complexes are unstable and decompose upon storage for a prolonged time. Only recently have copper(I) complexes of silylenes been explored in copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction (click reaction).89 The reactions of amidinato silylenes with CuX (X = Br, I, SCN) in a 1:1 molar ratio resulted in the formation of dinuclear copper complexes 99–101 (Scheme 38).90
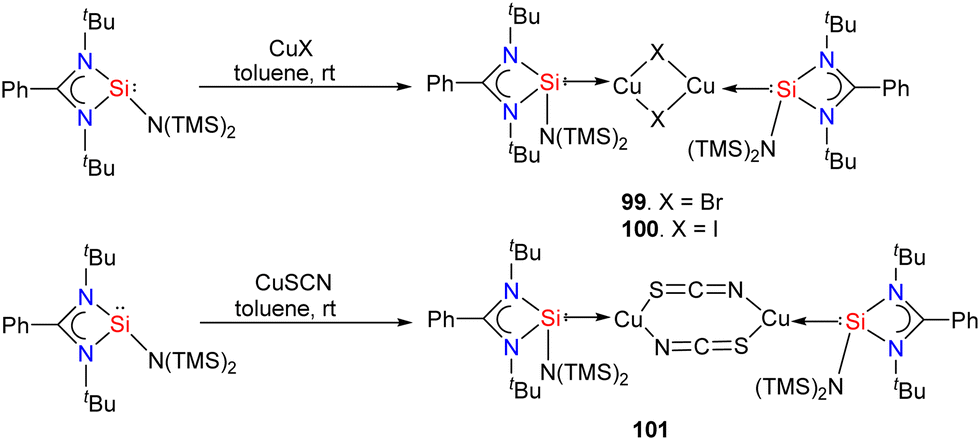 | ||
| Scheme 38 Synthesis of Cu(I) complexes 99–101. | ||
Further, the complexes were utilized in the catalytic copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. The copper complex 100 was the best catalyst, providing good to excellent yields of 1,2,3-triazoles with just 0.5 mol% of catalyst loadings. Various alkynes with electron-releasing and electron-withdrawing groups and organic azides were employed under the optimized reaction condition to afford good to excellent yield of the 1,2,3-triazoles (Scheme 39). Based on DFT studies, the authors proposed that the bimetallic Cu complexes act as active catalysts and accelerate the cycloaddition of reactants by bringing them nearby.
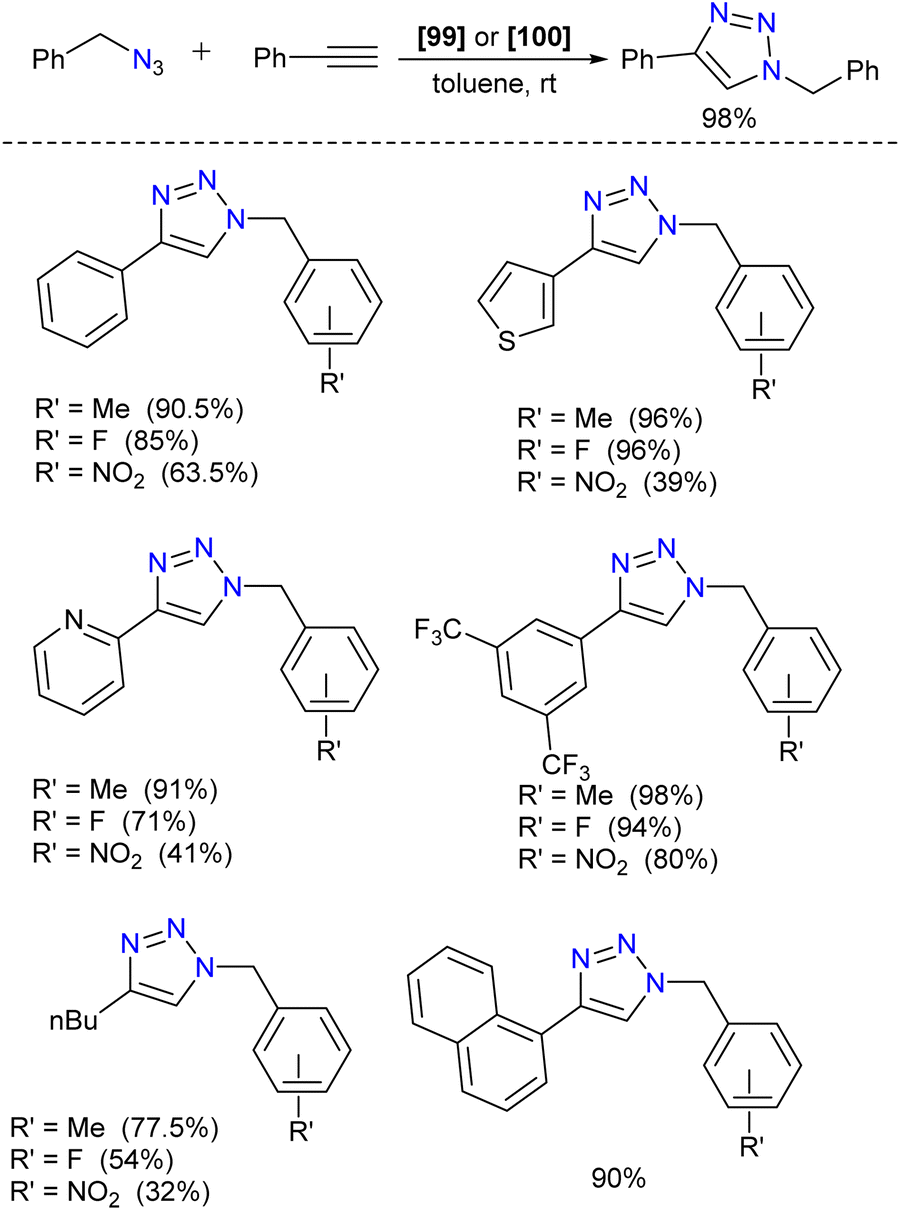 | ||
| Scheme 39 Substrate scope of CuAAC reaction. | ||
In 2022, Iwamoto and co-workers reported a series of neutral coinage metal complexes of cyclic alkylsilylene 102 and cyclic alkylaminosilylene 103.91 The reaction of 102 and 103 with 0.5 equivalents of MCl (M = Cu, Ag) or AuCl·tht (tht = tetrahydrothiopene) salts resulted in the formation of two coordinate neutral complexes 104M–105M (Scheme 40). Interestingly, during reactions with MCl salts, one equivalent of silylene ligands reacts via M–Cl bond insertion, and another equivalent of ligand coordinates to the metal center to give two coordinate neutral complexes. Remarkably, the Cu and Ag complexes exhibited a 1,3-Cl migration in solution states, as confirmed by a variable temperature 29Si NMR data suggesting that the 1,3-Cl migration follows the order Au < Ag < Cu and 104 < 105. The DFT calculations and X-ray structures suggest that the more bent structure of Cu and Ag over Au determines the relative ease of the 1,3-Cl migration.
 | ||
| Scheme 40 Synthesis of 104M–105M. | ||
Zn–silylene complexes
In recent years, significant attention has been devoted to the synthesis of organozinc compounds with N-heterocyclic carbenes, driven by their potential applications in organic catalysis. However, comparable reactions involving their heavier analogues (silylenes) are relatively scarce in the literature. In an attempt to capitalize on the synergistic reactivity effects of heterobimetallic hydride complexes containing main-group and transition metals, Schulz and co-workers conjectured that reactions of organozinc hydrides with silylenes might lead to the oxidative activation of Zn–H bonds at the low-valent Si center, affording novel heterobimetallic hydride complexes (Scheme 41).92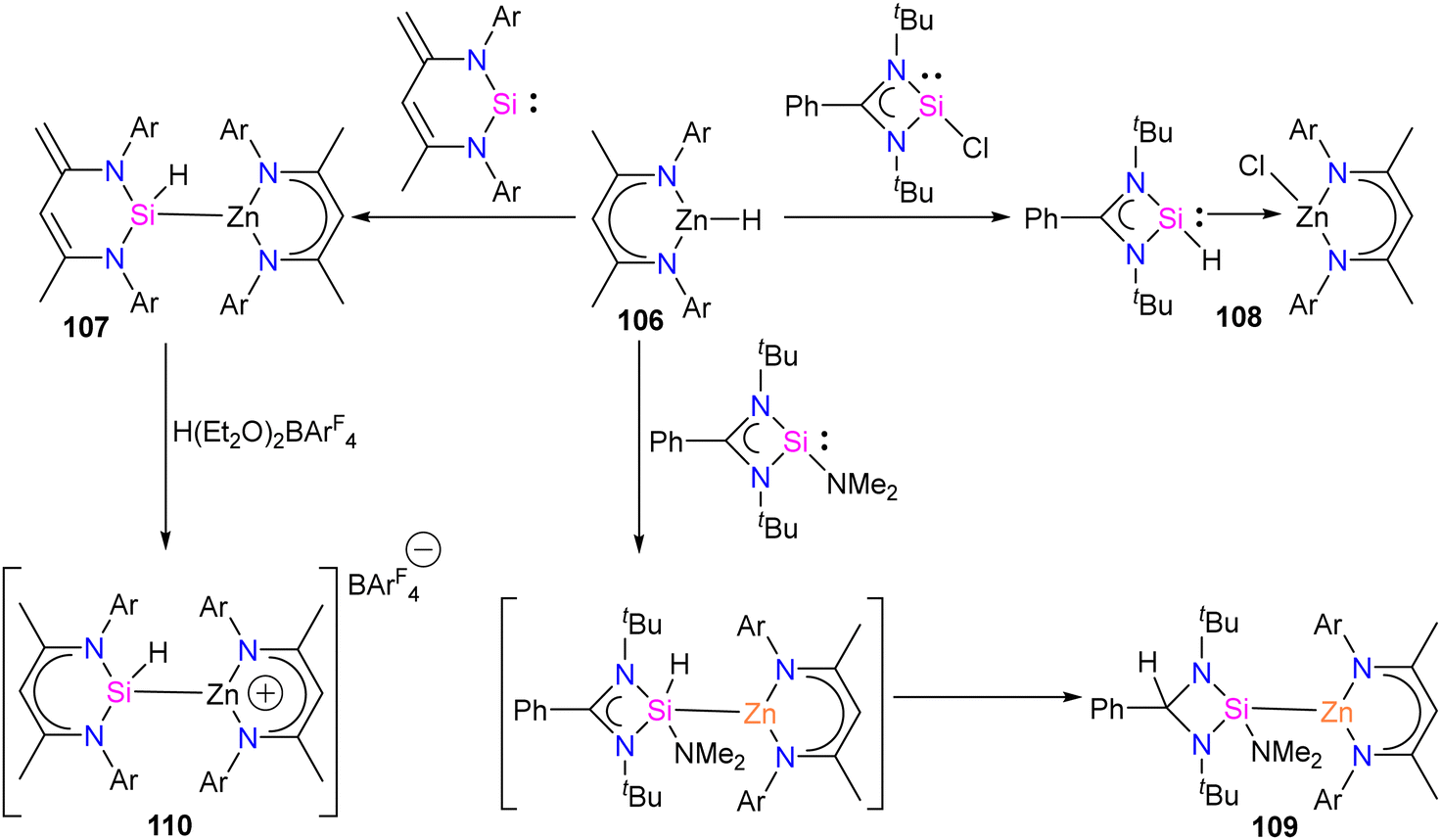 | ||
| Scheme 41 The reaction of organozinc hydride 106 with silylenes. | ||
The reaction of the LZnH complex 106 with N-ylidic silylene afforded the hypothesized Zn–H bond activation by silylene to afford heterobimetallic hydride complex 107. A similar reaction of heteroleptic chlorosilylene with LZnH proceeded through Cl/H exchange at the silicon center to afford complex 108. In sharp contrast, LZnH did not react with bulky L2SiN(SiMe3)2 even at high temperatures. Nevertheless, at 130 °C, the less bulky L2SiNMe2 reacted with LZnH, resulting in the oxidative addition of the Zn–H bond and a 1,3-H shift of the most likely formed silane reaction intermediates to LH2Si(NMe2)ZnL 109. This marked difference in reactivity was attributed to the stabilization/destabilization effect of electron-withdrawing/releasing substituent at the Zn atom. In the case of L2SiNMe2, the NMe2 group at the Si atom prevents the ligand exchange to form an adduct like 108. However, the bulky N(SiMe3)2 substituent prevents the oxidative addition, while the sterically less bulky NMe2 substituent permits the Zn–H bond to be added to the silylene in a 1,3-double bond. Further, the authors also tested the reactivity of 107 with isocyanates, azides, and CO2, but no reaction has occurred. Interestingly, complex 107 on treatment with [H(OEt2)2][BAr4F] (BAr4F = B{(3,5-CF3)2C6H3}4) resulted in the protonation of the methylene group of the ligand backbone, yielding the salt [LSi(H)ZnL]–[BAr4F] (110). The authors discovered that the protonation of the methylene group of ligand backbone is kinetically favored due to the weakly basic and less hindered methylene group. However, the 1,1-addition is unfavorable for the four-coordinated Si atom with a strong Si–Zn bond. According to the DFT calculation, complexes 107 and 109 have covalent Si–Zn bonds, while complex 108 has dative-type bonds.
Summary and outlook
In conclusion, this review discusses the most recent developments in the field of transition metal complexes of silylenes and their catalytic utility. The journeys through silylene chemistry underscores the transformative potential of tailored ligands in advancing various fields of chemistry, including main group chemistry, organometallic chemistry, catalysis and material chemistry. The evolution from unstable, reactive intermediates to stable and functionalized silylenes has opened new avenues for exploration and application. Recent years have seen utilization of transition metal complexes of silylenes in various important applications such valence tautomerism, small molecule activation chemistry, hydrogenation, hydroboration reactions and hydrogen isotope exchange reactions.While significant strides have been made, transition metal complexes of silylenes still lag behind NHC and phosphine-stabilized transition metal complexes in various important applications. The synthesis of high valent transition metal complexes of silylenes remains largely unexplored. The limitations in current synthetic techniques and the inherent instability of silylenes under oxidative condition necessitate a logical approach to develop innovative strategies for their preparation and utilization in catalytic processes.
Despite these challenges, the prospects are promising, with recent advancements showcasing the reactivity and versatility of silylene ligands, particularly in mimicking transition metal behaviour in small molecule activation chemistry. The ongoing pursuit of transition metal complexes of silylenes promises to unravel new insights and opportunities in catalysis and beyond.
Data availability
This is a review article. No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- (a) M. Stradiotto and R. J. Lundgren, Ligand Design in Metal Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2016 CrossRef; (b) D. J. Durand and N. Fey, Chem. Rev., 2019, 119, 6561–6594 CrossRef CAS PubMed; (c) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- (a) G. R. F. Orton, B. S. Pilgrim and N. R. Champness, Chem. Soc. Rev., 2021, 50, 4411–4431 RSC; (b) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119–2176 CrossRef PubMed; (c) C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116, 9237–9304 CrossRef CAS PubMed.
- (a) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (b) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- R. Schwarz and G. Pietsch, Z. Anorg. Allg. Chem., 1937, 232, 249–256 CrossRef CAS.
- T. J. Drahnak, J. Michl and R. West, J. Am. Chem. Soc., 1979, 101, 5427–5428 CrossRef CAS.
- M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- P. Jutzi, D. Kanne and C. Krüger, Angew. Chem., Int. Ed. Engl., 1986, 25, 164 CrossRef.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- N. J. Hill and R. West, J. Organomet. Chem., 2004, 689, 4165–4183 CrossRef CAS.
- (a) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS PubMed; (b) C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- (a) R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS PubMed; (b) A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 5687–5690 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, A. Kostenko and M. Driess, Dalton Trans., 2018, 47, 2152–2155 RSC.
- A. Hinz, Angew. Chem., Int. Ed., 2020, 59, 19065–19069 CrossRef CAS PubMed.
- S. Takahashi, J. Sekiguchi, A. Ishii and N. Nakata, Angew. Chem., Int. Ed., 2021, 60, 4055–4059 CrossRef CAS PubMed.
- (a) S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536–8538 CrossRef CAS PubMed; (b) S. S. Sen, S. Khan, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2012, 45, 578–587 CrossRef CAS PubMed; (c) S. S. Sen, S. Khan, P. P. Samuel and H. W. Roesky, Chem. Sci., 2012, 3, 659–682 RSC; (d) M. Haaf, A. Schmiedl, T. A. Schmedake, D. R. Powell, A. J. Millevolte, M. Denk and R. West, J. Am. Chem. Soc., 1998, 120, 12714–12719 CrossRef CAS.
- (a) M. Ghosh and S. Khan, Dalton Trans., 2021, 50, 10674–10688 RSC; (b) W. Yang, Y. Dong, H. Sun and X. Li, Dalton Trans., 2021, 50, 6766–6772 RSC; (c) C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC; (d) B. Blom, M. Stoelzel and M. Driess, Chem. – Eur. J., 2013, 19, 40–62 CrossRef CAS PubMed.
- (a) S. Fujimori and S. Inoue, Eur. J. Inorg. Chem., 2020, 3131–3142 CrossRef CAS PubMed; (b) M. Driess, Nat. Chem., 2012, 4, 525–526 CrossRef CAS PubMed; (c) J. Keuter, A. Hepp, A. Massolle, J. Neugebauer, C. Mück-Lichtenfeld and F. Lips, Angew. Chem., Int. Ed., 2022, 61, e202114485 CrossRef CAS PubMed.
- (a) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed; (b) Z. Jin, F. Zhang, X. Xiao, N. Wang, X. Lv and L. Zhou, Org. Chem. Front., 2024, 11, 2112–2133 RSC; (c) S. Kaufhold, L. Petermann, R. Staehle and S. Rau, Coord. Chem. Rev., 2015, 304–305, 73–87 CrossRef CAS; (d) M. Huang, J. Liu, Y. Li, X.-B. Lan, P. Su, C. Zhao and Z. Ke, Catal. Today, 2021, 370, 114–141 CrossRef CAS; (e) G. P. Mitchell and T. D. Tilley, Angew. Chem. Int. Ed., 1998, 37, 2524–2526 CrossRef CAS; (f) J. C. Peters, J. D. Feldman and T. D. Tilley, J. Am. Chem. Soc., 1999, 121, 9871–9872 CrossRef CAS.
- G. Schmid and E. Welz, Angew. Chem., Int. Ed. Engl., 1977, 16, 785–786 CrossRef.
- (a) C. Zybill and G. Müller, Angew. Chem., Int. Ed. Engl., 1987, 26, 669–670 CrossRef; (b) D. A. Straus, T. D. Tilley, A. L. Rheingold and S. J. Geib, J. Am. Chem. Soc., 1987, 109, 5872–5873 CrossRef CAS; (c) D. A. Straus, C. Zhang, G. E. Quimbita, S. D. Grumbine, R. H. Heyn, T. D. Tilley, A. L. Rheingold and S. J. Geib, J. Am. Chem. Soc., 1990, 112, 2673–2681 CrossRef CAS; (d) D. A. Straus, S. D. Grumbine and T. D. Tilley, J. Am. Chem. Soc., 1990, 112, 7801–7802 CrossRef CAS.
- (a) B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134–148 RSC; (b) S. Yao, A. Saddington, Y. Xiong and M. Driess, Acc. Chem. Res., 2023, 56, 475–488 CrossRef CAS PubMed; (c) S. Raoufmoghaddam, Y.-P. Zhou, Y. Wang and M. Driess, J. Organomet. Chem., 2017, 829, 2–10 CrossRef CAS.
- T. A. Schmedake, M. Haaf, B. J. Paradise, A. J. Millevolte, D. R. Powell and R. West, J. Organomet. Chem., 2001, 636, 17–25 CrossRef CAS.
- W. Yang, H. Fu, H. Wang, M. Chen, Y. Ding, H. W. Roesky and A. Jana, Inorg. Chem., 2009, 48, 5058–5060 CrossRef CAS PubMed.
- (a) X. Du, X. Qi, K. Li, X. Li, H. Sun, O. Fuhr and D. Fenske, Appl. Organomet. Chem., 2021, 35, e6286 CrossRef CAS; (b) S. Khoo, J. Cao, F. Ng and C.-W. So, Inorg. Chem., 2018, 57, 12452–12455 CrossRef CAS PubMed.
- Y. Bai, J. Zhang and C. Cui, Chem. Commun., 2018, 54, 8124–8127 RSC.
- R. Azhakar, S. P. Sarish, H. W. Roesky, J. Hey and D. Stalke, Inorg. Chem., 2011, 50, 5039–5043 CrossRef CAS PubMed.
- S. Fujimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS PubMed.
- P. Garg, A. Carpentier, I. Douair, D. Dange, Y. Jiang, K. Yuvaraj, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2022, 61, e202201705 CrossRef CAS PubMed.
- K. Ogata, Y. Yamaguchi, Y. Kurihara, K. Ueda, H. Nagao and T. Ito, Inorg. Chim. Acta, 2012, 390, 199–209 CrossRef CAS.
- (a) S. Khoo, J. Cao, M.-C. Yang, Y.-L. Shan, M.-D. Su and C.-W. So, Chem. – Eur. J., 2018, 24, 14329–14334 CrossRef CAS PubMed; (b) S. Khoo, H.-X. Yeong, Y. Li, R. Ganguly and C.-W. So, Inorg. Chem., 2015, 54, 9968–9975 CrossRef CAS PubMed.
- Z. He, L. Liu, F. J. de Zwart, X. Xue, A. W. Ehlers, K. Yan, S. Demeshko, J. I. van der Vlugt, B. de Bruin and J. Krogman, Inorg. Chem., 2022, 61, 11725–11733 CrossRef CAS PubMed.
- M. Ichinohe, M. Igarashi, K. Sanuki and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 9978–9979 CrossRef CAS PubMed.
- S. S. Sen, G. Tavčar, H. W. Roesky, D. Kratzert, J. Hey and D. Stalke, Organometallics, 2010, 29, 2343–2347 CrossRef CAS.
- R. Akhtar, S. H. Kaulage, M. P. Sangole, S. Tothadi, P. Parvathy, P. Parameswaran, K. Singh and S. Khan, Inorg. Chem., 2022, 61, 13330–13341 CrossRef CAS PubMed.
- A. Saurwein, T. Eisner, S. Inoue and B. Rieger, Organometallics, 2022, 41, 3679–3685 CrossRef CAS.
- F. Masero, M. A. Perrin, S. Dey and V. Mougel, Chem. – Eur. J., 2021, 27, 3892–3928 CrossRef CAS PubMed.
- C. Sivasankar, P. K. Madarasi and M. Tamizmani, Eur. J. Inorg. Chem., 2020, 1383–1395 CrossRef CAS.
- W. Yang, X. Li, S.-Y. Li, Q. Li, H. Sun and X. Li, Inorg. Chem., 2023, 62, 21014–21024 CrossRef CAS PubMed.
- S. K. Kushvaha, P. Kallenbach, S. M. N. V. T. Gorantla, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Chem. – Eur. J., 2024, 30, e202303113 CrossRef CAS PubMed.
- M. K. Pandey, Z. Hendi, X. Wang, A. Bhandari, M. K. Singh, K. Rachuy, S. Kumar Kushvaha, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Angew. Chem., Int. Ed., 2023, 63, e202317416 CrossRef PubMed.
- R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Organometallics, 2012, 31, 4588–4592 CrossRef CAS.
- Z. Hendi, M. K. Pandey, K. Rachuy, M. K. Singh, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Chem. – Eur. J., 2024, 30, e202400389 CrossRef CAS PubMed.
- (a) L. A. Saudan, Acc. Chem. Res., 2007, 40, 1309–1319 CrossRef CAS PubMed; (b) B. Chen, U. Dingerdissen, J. G. E. Krauter, H. G. J. Lansink Rotgerink, K. Möbus, D. J. Ostgard, P. Panster, T. H. Riermeier, S. Seebald, T. Tacke and H. Trauthwein, Appl. Catal., A, 2005, 280, 17–46 CrossRef CAS.
- Y. Wang, A. Kostenko, S. Yao and M. Driess, J. Am. Chem. Soc., 2017, 139, 13499–13506 CrossRef CAS PubMed.
- H. Jia, S. Du, C. Xu and Z. Mo, Eur. J. Inorg. Chem., 2023, e202300086 CrossRef CAS.
- Q. Fan, X. Du, W. Yang, Q. Li, W. Huang, H. Sun, A. Hinz and X. Li, Dalton Trans., 2023, 52, 6712–6721 RSC.
- X. Qi, H. Sun, X. Li, O. Fuhr and D. Fenske, Dalton Trans., 2018, 47, 2581–2588 RSC.
- (a) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed; (b) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- (a) J. T. Golden, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 2001, 123, 5837–5838 CrossRef CAS PubMed; (b) M. H. G. Prechtl, M. Hölscher, Y. Ben-David, N. Theyssen, R. Loschen, D. Milstein and W. Leitner, Angew. Chem., Int. Ed., 2007, 46, 2269–2272 CrossRef CAS PubMed.
- J. Corpas, P. Viereck and P. J. Chirik, ACS Catal., 2020, 10, 8640–8647 CrossRef CAS.
- S. Garhwal, A. Kaushansky, N. Fridman, L. J. W. Shimon and G. d Ruiter, J. Am. Chem. Soc., 2020, 142, 17131–17139 CrossRef CAS PubMed.
- A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478–11482 CrossRef PubMed.
- R. Arevalo, T. P. Pabst and P. J. Chirik, Organometallics, 2020, 39, 2763–2773 CrossRef CAS PubMed.
- J. B. Roque, T. P. Pabst and P. J. Chirik, ACS Catal., 2022, 12, 8877–8885 CrossRef CAS PubMed.
- S. P. Church, M. Poliakoff, J. A. Timney and J. J. Turner, J. Am. Chem. Soc., 1981, 103, 7515–7520 CrossRef CAS.
- T. T. Metsänen, D. Gallego, T. Szilvási, M. Driess and M. Oestreich, Chem. Sci., 2015, 6, 7143–7149 RSC.
- S. Kalra, D. Pividori, D. Fehn, C. Dai, S. Dong, S. Yao, J. Zhu, K. Meyer and M. Driess, Chem. Sci., 2022, 13, 8634–8641 RSC.
- M. Rotter, M. Mastalir, M. Glatz, B. Stoger and K. Kirchner, Acta Crystallogr., 2017, 73, 1308–1311 CrossRef CAS PubMed.
- D. Reardon, G. Aharonian, S. Gambarotta and G. P. A. Yap, Organometallics, 2002, 21, 786–788 CrossRef CAS.
- D. W. Agnew, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Angew. Chem., Int. Ed., 2015, 54, 12673–12677 CrossRef CAS PubMed.
- H. Ahuja, H. Kaur and R. Arevalo, Inorg. Chem. Front., 2023, 10, 6067–6076 RSC.
- M. Denk, R. K. Hayashi and R. West, J. Chem. Soc., Chem. Commun., 1994, 33–34 RSC.
- B. Gehrhus, P. B. Hitchcock, M. F. Lappert and H. Maciejewski, Organometallics, 1998, 17, 5599–5601 CrossRef CAS.
- T. J. Hadlington, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 7470–7474 CrossRef CAS PubMed.
- T. J. Hadlington, A. Kostenko and M. Driess, Chem. – Eur. J., 2020, 26, 1958–1962 CrossRef CAS PubMed.
- M. Zhong, J. Wei, W.-X. Zhang and Z. Xi, Organometallics, 2021, 40, 310–313 CrossRef CAS.
- D. F. Shriver, Acc. Chem. Res., 1970, 3, 231–238 CrossRef CAS.
- (a) D. M. T. Chan and T. B. Marder, Angew. Chem., Int. Ed. Engl., 1988, 27, 442–443 CrossRef; (b) C. Gendy, A. Mansikkamäki, J. Valjus, J. Heidebrecht, P. C.-Y. Hui, G. M. Bernard, H. M. Tuononen, R. E. Wasylishen, V. K. Michaelis and R. Roesler, Angew. Chem., Int. Ed., 2019, 58, 154–158 CrossRef CAS PubMed.
- M. Frutos, N. Parvin, A. Baceiredo, D. Madec, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2022, 61, e202201932 CrossRef CAS PubMed.
- (a) A. Zeller, F. Bielert, P. Haerter, W. A. Herrmann and T. Strassner, J. Organomet. Chem., 2005, 690, 3292–3299 CrossRef CAS; (b) C. Watanabe, Y. Inagawa, T. Iwamoto and M. Kira, Dalton Trans., 2010, 39, 9414–9420 RSC; (c) G. P. Mitchell and T. D. Tilley, Angew. Chem., Int. Ed., 1998, 37, 2524–2526 CrossRef CAS; (d) S. D. Grumbine, T. D. Tilley, A. L. Rheingold and F. P. Arnold, J. Am. Chem. Soc., 1993, 115, 7884–7885 CrossRef CAS; (e) J. D. Feldman, G. P. Mitchell, J.-O. Nolte and T. D. Tilley, J. Am. Chem. Soc., 1998, 120, 11184–11185 CrossRef CAS; (f) J. D. Feldman, G. P. Mitchell, J. O. Nolte and T. D. Tilley, Can. J. Chem., 2003, 81, 1127–1136 CrossRef CAS; (g) C. Watanabe, T. Iwamoto, C. Kabuto and M. Kira, Angew. Chem., Int. Ed., 2008, 47, 5386–5389 CrossRef CAS PubMed; (h) C. Watanabe, Y. Inagawa, T. Iwamoto and M. Kira, Dalton Trans., 2010, 39, 9414–9420 RSC; (i) K. Osakada, M. Tanabe and T. Tanase, Angew. Chem., Int. Ed., 2000, 39, 4053–4055 CrossRef CAS; (j) Y. Sunada, N. Taniyama, K. Shimamoto, S. Kyushin and H. Nagashima, Inorganics, 2017, 5, 84 CrossRef.
- (a) T. D. Tilley, Comments Inorg. Chem., 1990, 10, 37–51 CrossRef CAS; (b) K. Yamamoto, H. Okinoshima and M. Kumada, J. Organomet. Chem., 1970, 23, C7–C8 CrossRef CAS; (c) K. Yamamoto, T. Hayashi and M. Kumada, J. Organomet. Chem., 1971, 28, C37–C38 CrossRef CAS.
- A. G. Avent, B. Gehrhus, P. B. Hitchcock, M. F. Lappert and H. Maciejewski, J. Organomet. Chem., 2003, 686, 321–331 CrossRef CAS.
- P. Hädinger and A. Hinz, Dalton Trans., 2023, 52, 2214–2218 RSC.
- M. Tanabe, Y. Nakamura, T. A. Niwa, M. Sakai, A. Kaneko, H. Toi, K. Okuma, Y. Tsuchido, T. A. Koizumi, K. Osakada and T. Ide, Organometallics, 2022, 41, 3301–3312 CrossRef CAS.
- J. Sekiguchi, Y. Kazama, A. Ishii and N. Nakata, Chem. Commun., 2023, 59, 9844–9847 RSC.
- G. Tan, B. Blom, D. Gallego and M. Driess, Organometallics, 2014, 33, 363–369 CrossRef CAS.
- P. Jutzi and A. Möhrke, Angew. Chem., Int. Ed. Engl., 1990, 29, 893–894 CrossRef.
- A. G. Avent, B. Gehrhus, P. B. Hitchcock, M. F. Lappert and H. Maciejewski, J. Organomet. Chem., 2003, 686, 321–331 CrossRef CAS.
- C. G. Pierpont, Coord. Chem. Rev., 2001, 216–217, 99–125 CrossRef CAS.
- L. Greb, Eur. J. Inorg. Chem., 2022, e202100871 CrossRef CAS PubMed.
- S. Yao, A. Kostenko, Y. Xiong, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2020, 142, 12608–12612 CrossRef CAS PubMed.
- T. Koike, T. Nukazawa and T. Iwamoto, J. Am. Chem. Soc., 2021, 143, 14332–14341 CrossRef CAS PubMed.
- R. Yadav, X. Sun, R. Köppe, M. T. Gamer, F. Weigend and P. W. Roesky, Angew. Chem., Int. Ed., 2022, 61, e202211115 CrossRef CAS PubMed.
- (a) T. Wurm, A. Mohamed Asiri and A. S. K. Hashmi, N-Heterocyclic Carbenes, 2014, pp. 243–270 DOI:10.1002/9783527671229.ch09; (b) W. Zi and F. Dean Toste, Chem. Soc. Rev., 2016, 45, 4567–4589 RSC; (c) V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC; (d) Z. Hendi, S. Jamali, S. M. J. Chabok, A. Jamjah, H. Samouei and Z. Jamshidi, Inorg. Chem., 2021, 60, 12924–12933 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- M. Nazish, H. Bai, C. M. Legendre, R. Herbst-Irmer, L. Zhao, D. Stalke and H. W. Roesky, Chem. Commun., 2022, 58, 12704–12707 RSC.
- Y.-P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed.
- (a) A. N. Paesch, A.-K. Kreyenschmidt, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2019, 58, 7000–7009 CrossRef CAS PubMed; (b) N. Parvin, J. Hossain, A. George, P. Parameswaran and S. Khan, Chem. Commun., 2020, 56, 273–276 RSC.
- J. Hossain, J. S. Gopinath, S. Tothadi, P. Parameswaran and S. Khan, Organometallics, 2022, 41, 3706–3717 CrossRef CAS.
- S. Abe, Y. Inagawa, R. Kobayashi, S. Ishida and T. Iwamoto, Organometallics, 2022, 41, 874–882 CrossRef CAS.
- B. Li, H. M. Weinert, C. Wölper and S. Schulz, Organometallics, 2023, 42, 457–464 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |