
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating iminophosphorane P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N bond reactivity through geometric constraints with cage-shaped triarylphosphines†
N bond reactivity through geometric constraints with cage-shaped triarylphosphines†
Lei
Hu‡§
ab,
Sayandip
Chakraborty§
a,
Nikolay
Tumanov
 a,
Johan
Wouters
a,
Raphaël
Robiette
*b and
Guillaume
Berionni
*a
a,
Johan
Wouters
a,
Raphaël
Robiette
*b and
Guillaume
Berionni
*a
aUniversité de Namur, Department of Chemistry, Namur Institute of Structured Matter (NISM), Rue de Bruxelles 61, Namur 5000, Belgium. E-mail: guillaume.berionni@unamur.be
bUniversité Catholique de Louvain, Institute of Condensed Matter and Nanosciences, Place Louis Pasteur 1 box L4.01.02, Louvain-la-Neuve 1348, Belgium. E-mail: raphael.robiette@uclouvain.be
First published on 18th June 2024
Abstract
Structure–reactivity investigations and quantum-chemical parametrization of steric and electronic properties of geometrically constrained iminophosphoranes enabled the design of new frustrated Lewis pairs and revealed unusual properties at the phosphonium center embedded in the cage-shaped triptycene tricyclic scaffold.
Iminophosphoranes, the nitrogen analogues of phosphorus ylides in aza-Wittig reactions,1 are increasingly used to design pincer type ligands for transition-metals2 and main-group elements.3 They have found recent applications in organocatalysis4 and in the phosphine-mediated redox catalyzed Staudinger ligation of carboxylic acids and azides (Scheme 1a).5 Iminophosphoranes are prepared by the Staudinger reaction,1a whose reaction intermediates have been extensively investigated (Scheme 1b).6
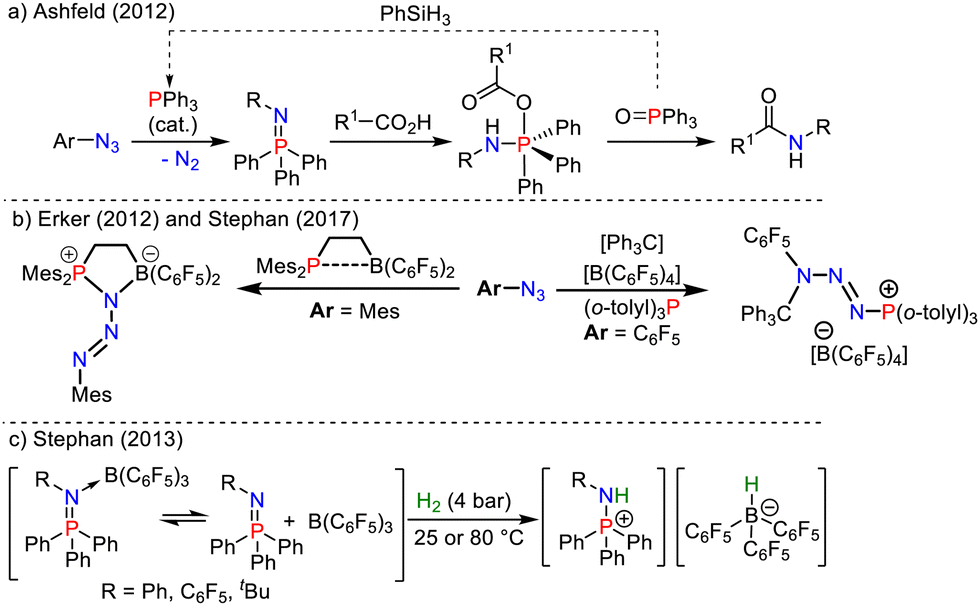 | ||
| Scheme 1 (a) PIII/PV-redox catalyzed Staudinger ligation reaction.5 (b) Interrupted Staudinger reactions.6 (c) Applications of iminophosphoranes in FLP chemistry.8 | ||
Owing to the high basicity of their nitrogen atom, imino-phosphoranes have been extensively used to design superbases (e.g. Schwesinger phosphazenes),7 and have been combined with bulky boron Lewis acids such as B(C6F5)3 to generate frustrated Lewis pairs (FLPs) reacting with small molecules such as CO2 and H2 (Scheme 1c).8
Whereas the reactivity of these phospha-aza ylides is usually governed by the electronic and steric properties of the substituents at the P and N atoms, constraining their geometry with a highly strained skeleton or cage-shaped framework is of increasing interest for modulating the reactivity of p-block compounds and reaching new reactivities (Scheme 2).9
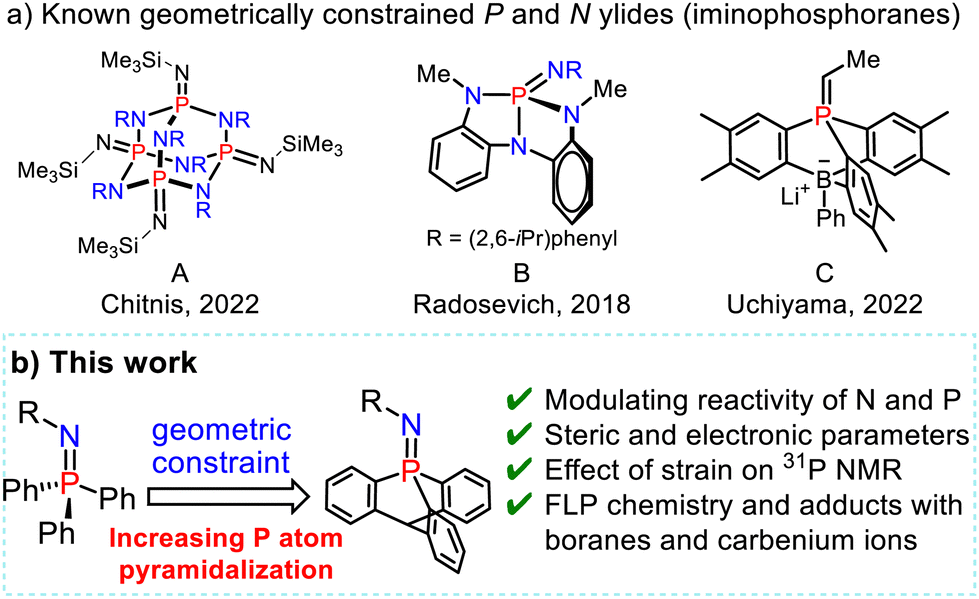 | ||
Scheme 2 (a) Examples of geometrically constrained phosphorus frameworks. (b) This work: exploiting geometric distortion at phosphorus to modulate the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond reactivity and steric hindrance. N bond reactivity and steric hindrance. | ||
Chitnis and coworkers recently designed phosphaza-adamantane with thermal, air, and redox stability by using a geometrically constrained adamantane scaffold (Scheme 2a-A).10 Radosevich and coworkers modulated the properties of the PN bond in phosphazenes by designing phosphabicyclic compounds with distorted T-shaped molecular geometries, enabling the tuning of their reactivity via a geometric constraint (Scheme 2a-B).11 Uchiyama recently employed a phospha-boratriptycene cage-shaped scaffold to intercept betaine intermediates in Wittig reactions of phosphorus ylides with aldehydes (Scheme 2a-c).12
We now report a series of phosphatricyclic nitrogen-ylides derived from 9-phosphatriptycene,13 in which the geometric constraint is inducing an unusual pyramidalized phosphorus environment at the edge of the triptycene scaffold.14 The impact of the geometrical constraint on the Lewis and Brønsted basicities, steric hindrance and reactivity of the PN bond was elucidated by quantitative investigations of their association with carbenium ions, and Brønsted and Lewis acids.
Adducts featuring unprecedented non-covalent fluorine or oxygen non-covalent bonding to C–P σ* orbitals were obtained and the striking reactivities of the cage-shaped phosphinimines were further exploited to design FLPs with bulky Lewis acids.
We first compared the magnetic and structural properties of triphenylphosphine 2–4 and triptycene chalcogenides 6–8 (Table 1). Single-crystal X-ray diffraction analysis revealed that TripPE derivatives 6–8 exhibited a larger pyramidalization angle α than in classical Ar3PE derivatives 2–4, corresponding to larger CPC angles of 107.7° versus 97.7°. In 31P NMR spectroscopy, a large shielding is observed between the 9-phosphatriptycene 5 (−64.4 ppm) and Ph3P 1 (−4.7 ppm), while a smaller shielding was observed between Trip-PE and Ph3PE derivatives (Table 1). Consistent with the literature,14e the 1J31P–77Se of 822 Hz in 8 is nearly 100 Hz larger than for PPh3Se 4 (729 Hz) indicating that the overall donating ability of phosphatriptycene 5 is less than Ph3P.13a Compound 6 was found to stabilize a molecule of H2O2 (Fig. S1 in the ESI†).
| Chalcogenide derivatives with E = | O | S | Se | ||
|---|---|---|---|---|---|
| a Pyramidalization angle α defined as the angle between the C–P bonds and the plane formed by the ipso-carbon atoms of the triptycene aryl rings, see ESI. | |||||
| Triphenylphosphine | 1 | 2 | 3 | 4 | |
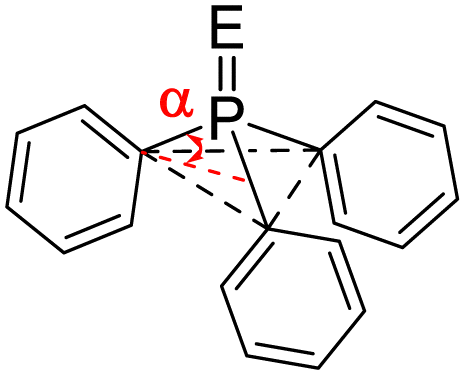
|
P–E | — | 1.482 (3) | 1.955 (7) | 2.114 (8) |
| α | 26.114a | 22.414b | 22.914d | 22.614g | |
| δ 31P ppm | −4.7 | 29.214c | 43.314e | 35.314f | |
| Phosphatriptycene | 5 | 6 | 7 | 8 | |
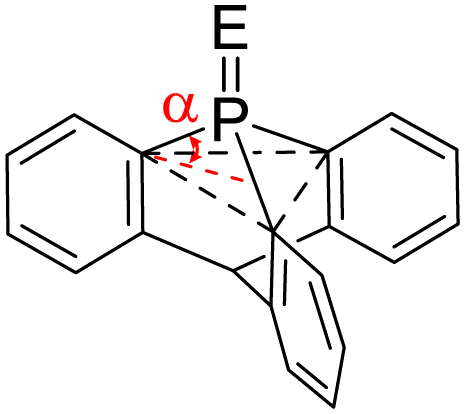
|
P–E | — | 1.482 (1) | 1.941 (1) | 2.091 (1) |
| α | 29.8 | 28.7 | 29.2 | 29.1 | |
| δ 31P ppm | −64.414h | 7.32 | 12.72 | 4.52 | |
We then performed the Staudinger reactions of 9-phosphatriptycene 5 with several azides R–N3 to synthesize the cage-shaped iminophosphoranes 9–14 (Scheme 3). According to the classical mechanism,15 these reactions are likely proceeding via an unusual inorganic spiro-cyclic transition-state with a four-membered PNNN ring connected to a phosphabicyclo[2.2.2]octane tricyclic core.
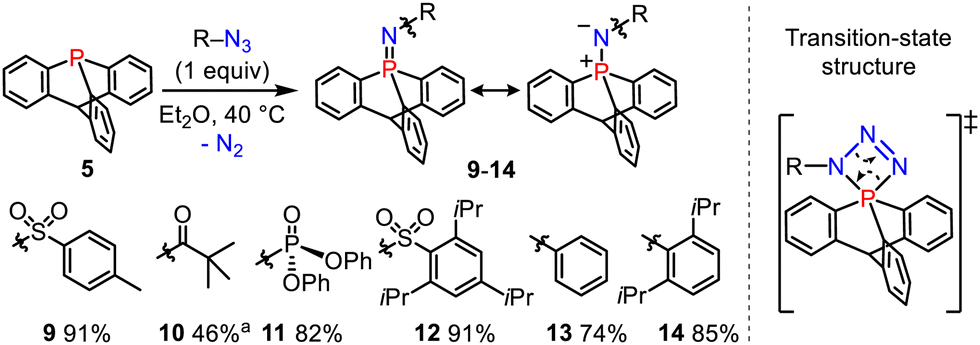 | ||
Scheme 3 Synthesis of the 9-phosphatriptycene-imines 9–14. a ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Prepared with different synthetic procedures, see the ESI† for more details. Prepared with different synthetic procedures, see the ESI† for more details. | ||
The 31P NMR chemical shifts of 9–14 (−5 to −33 ppm) were again more shielded than in the corresponding Ph3PNR derivatives reported in the literature (0 to 15 ppm).14 The formal PN double bond has predominantly an ylidic nature (see right-hand side resonance structure, Scheme 3) as confirmed by NBO calculations showing that the short PN bond lengths are resulting from negative hyperconjugation between the N lone pairs and 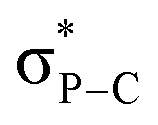 antibonding orbitals (2nd order perturbation stabilization energy E2 = 9.8, 33.2 and 33.3 kcal mol−1 for 13).16
antibonding orbitals (2nd order perturbation stabilization energy E2 = 9.8, 33.2 and 33.3 kcal mol−1 for 13).16
X-ray crystallographic analysis of compounds 9–14 (see ESI†) revealed a high pyramidalization angle at P, e.g. α = 29.0° in 13 and only 23.3° for its Ph3PNPh analogue (Fig. 1a and b).
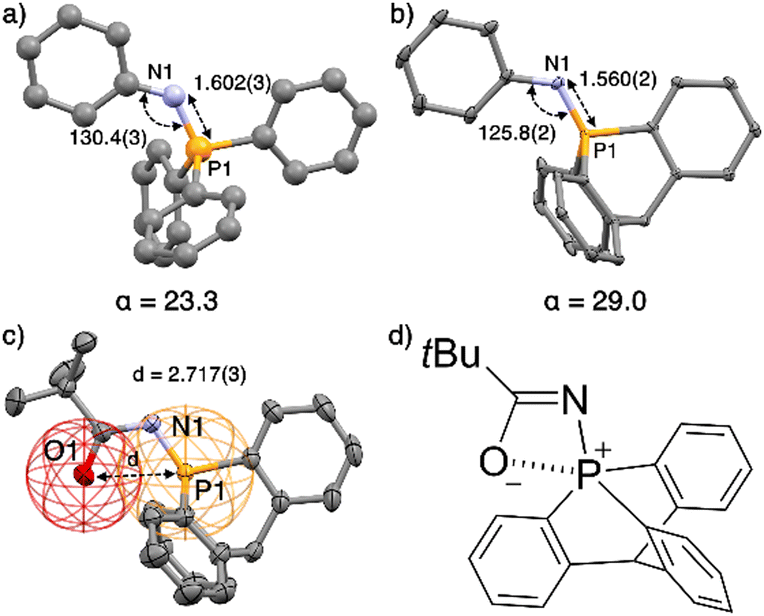 | ||
| Fig. 1 (a) Structure of Ph3PNPh17e,f and (b) of its analogue 13 with key geometrical features. (c) Ellipsoid representation of single crystal X-ray structures of 10; H atoms and solvent are omitted for clarity. (d) Preponderant mesomeric structure of 10. Distances in Å, angles in °, pyramidalization angle α in °. | ||
Compound 10 has a P⋯O distance (2.717(3) Å) well under the van der Waals radii of both atoms (represented by the red and yellow wireframe spheres around O and P, respectively, Fig. 1c), implying a preponderant resonance structure with marked electrostatic interaction between the positively charged P atom and negatively charged O atom (Fig. 1d).
This interaction is also confirmed by the NBO analysis of 10, which reveals an electronic interaction between one lone pair of the oxygen atom and one 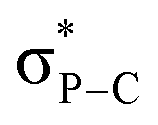 bond (E2 = 2.2 kcal mol−1). Due to the cage-shaped structure of phosphatriptycene, this P-distance is significantly shorter than in any previous reported phosphorus imidates (previous shortest = 2.848(2) Å for the corresponding Ph3PNCOtBu analogue of 10).17
bond (E2 = 2.2 kcal mol−1). Due to the cage-shaped structure of phosphatriptycene, this P-distance is significantly shorter than in any previous reported phosphorus imidates (previous shortest = 2.848(2) Å for the corresponding Ph3PNCOtBu analogue of 10).17
The iminophosphorane 13 was used for subsequent reactivity studies and was reacted with triflimidic acid HNTf2 and BF3·OEt2 giving respectively 15 and 16 in good yields (Scheme 4). Protonation and BF3 complexation induced a deshielding of the 31P NMR signals of 15 (10.7 ppm) and 16 (10.4 ppm) compared to that of 13 (−20.4 ppm). Crystallographic analysis showed that H+ and BF3 complexation of 13 resulted in a significant P–N lengthening, with a PN distance of 1.560(2) Å in 13, 1.615(1) Å in 15 and 1.611(2) Å in 16 (Fig. 2). This can be accounted for by the loss of one lone pair on the nitrogen that decreases the hyperconjugation with the 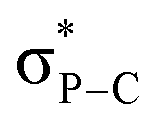 orbitals (E2 = 13.3 and 13.5 kcal mol−1 for 15) as compared to what was observed in 13 (vide supra).
orbitals (E2 = 13.3 and 13.5 kcal mol−1 for 15) as compared to what was observed in 13 (vide supra).
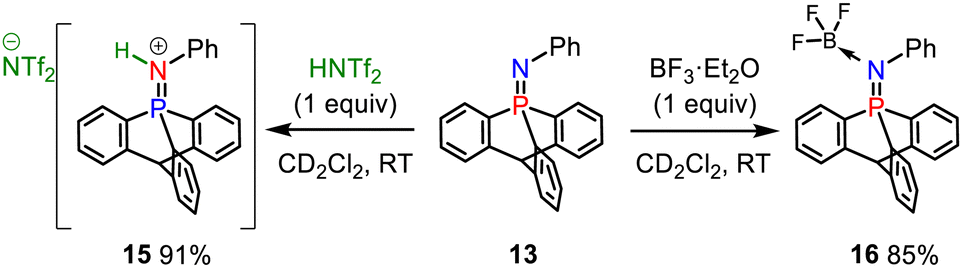 | ||
| Scheme 4 Reaction of 13 with a Brønsted and a boron Lewis acid. | ||
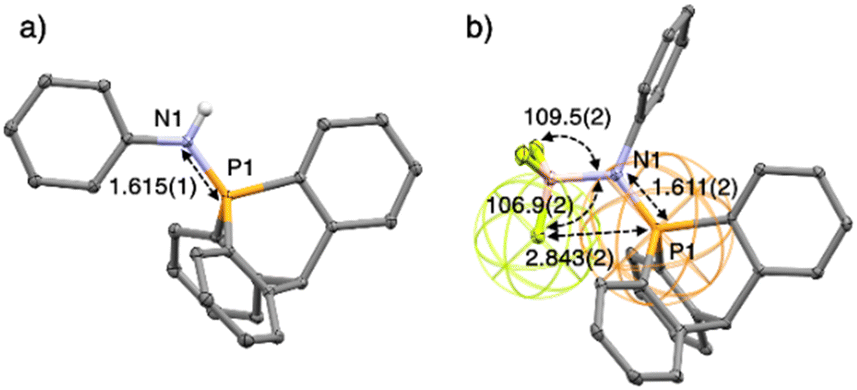 | ||
| Fig. 2 Ellipsoid representation (50% probability level) of single-crystal X-ray structures of one of the two molecules in the asymmetric unit of 15 (a), and 16 (b). H atoms, anion and solvent are omitted for clarity. | ||
The structure of 16 in the solid state revealed that one F atom in BF3 is anti (θC–P–F = 173.65(8)°) to one of the P–Carom bonds (Fig. 2b). The valence angle θN–B–F for F1 is 106.9(2)°, whereas it is 109.5(2)° and 109.8(2)° for the other two fluorine atoms. This suggests an interaction between the lone pair of one fluorine with the σ* orbital of one Carom–P bond. This P⋯F distance of 2.8433(14) Å is longer than in the phosphonium based anion receptors of Gabbaï (2.666(2) Å) in which the phosphonium bears a positive charge.18 Nevertheless, it is still very short with a F···P distance in 16, well below the sum of the van der Waals radii of both atoms (3.35 Å), represented by wireframe coloured spheres around F and P (Fig. 2b).
Then, the association of 13–14 with tris(pentafluorophenyl)-borane 17 was investigated by multinuclear NMR spectroscopy (Scheme 5). When mixing these reagents in CDCl3, a negligible deshielding of the signal of 13 (≈3 ppm) was observed by 31P NMR spectroscopy, and only a broadening of the peaks was observed in 1H NMR, while the 11B NMR signal of 17 remained at −60.0 ppm. This indicated a very weak interaction between 13 and B(C6F5)3 typical of an encounter complex or a frustrated Lewis pair, and the formation of a Lewis adduct was thus excluded. After keeping the FLP solution for one day, crystals of compound 18 were obtained (see ESI,† Fig. S2) among other decomposition products. Thus, the deactivation of FLP occurs partly via SNAr reaction of the nitrogen atom of the 9-phospatriptycene imine 13 at the para position of a C6F5 ring of B(C6F5)3 (Scheme 5), similarly as with phosphorus ylides.8c In the case of 14, since it is of larger steric hindrance around it's nitrogen atom, such type of SNAr reaction to yield 19 was not possible.
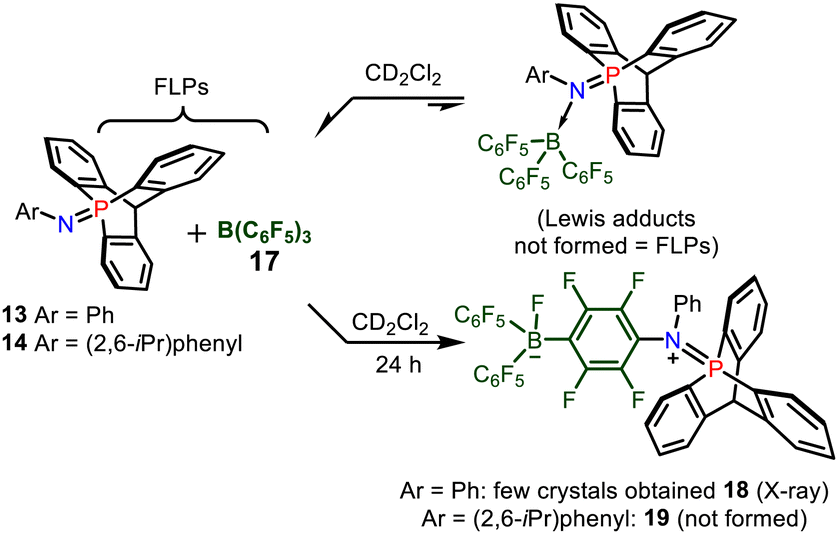 | ||
| Scheme 5 FLPs between 13 and 17 and deactivation pathway resulting in the formation of the phospha-iminium fluoroborate 18. | ||
The steric hindrance at the nitrogen atom was comparable for TripPNPh (13) and Ph3PNPh according to their similar buried volume %Vbur and He8_steric parameters (see ESI,† Table S3).19 Analogously, the %Vbur of phosphatriptycene 5 (31.3%) is comparable to that of Ph3P 1 (29.6%).20
Computations show that the proton (PA) and methyl cation affinities (MCA) of iminophosphoranes 9–14 are influenced by geometric constraints at the phosphorus atom. This constraint decreases the basicity at N by up to 8 kcal mol−1 when comparing 13 to is analogue Ph3PNPh (Table 2). It simultaneously enhances the Lewis acidity at the phosphorus atom by 4 kcal mol−1, as evidenced by fluoride ion affinity (FIA) values at the electron deficient formal phosphonium P centre. Analysis of the fluoride complexation through the activation strain model (see ESI,† Table S4) indicates that this enhanced Lewis acidity is mainly due to a lower distortion energy in the case of 13. The energy necessary for the geometrically constrained phosphatriptycene imine structure to adopt the trigonal bipyramidal geometry is indeed lower (by 4.6 kcal mol−1) than the one required for Ph3PNPh.
| Cmpd B | 9 | 10 | 11 | 12 | 13 | 14 | Ph3PNPh |
|
|---|---|---|---|---|---|---|---|---|
| a Computed FIA at the pseudo phosphonium center using the isodesmic approach with COF2 anchor points; see ESI, Table S2 for the FSiMe3 anchor point and for FIAs for 6–8. b In agreement with the reported FIA value for compound B (Radosevich T-shaped iminophosphoranes shown in Scheme 2b) of 57 kcal mol−1 in ref. 11. | ||||||||
| PA | 263 | 247 | 255 | 246 | 245 | 262 | 264 | 270 |
| MCA | 110 | 93 | 95 | 92 | 94 | 108 | 113 | 113 |
| FIAa | 60b | 47 | 37 | 45 | 51 | 36 | 38 | 32 |
Finally, we found that combining 13 with a titylium ion results in a nitrogen/carbon FLP, and the phospha-iminium 20 was not formed (Scheme 6), but instead SEAr reactions occurred at the para position of N-Ph of 13 to yield compound 21, evidenced by X-ray diffraction (Fig. S2b, ESI†).
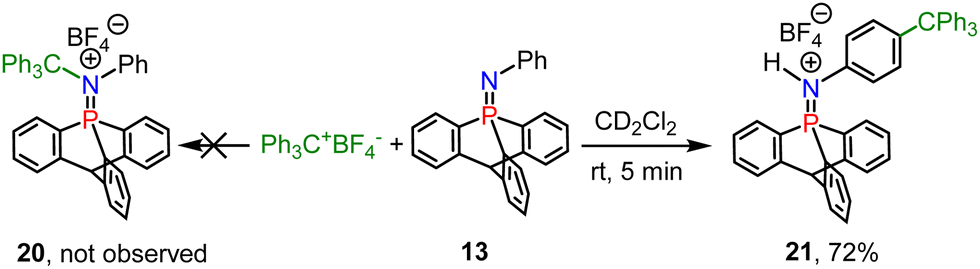 | ||
| Scheme 6 FLP type reaction of iminophosphorane 13 with the tritylium tetrafluoroborate to yield compound 21. | ||
Thus, in conclusion, the geometric constraints brought by the cage-shaped tricyclic phosphatriptycene scaffold decrease the Brønsted basicity at the nitrogen but enhance the Lewis acidity at the phosphorus, leading to a partial Umpolung type reactivity at the PN bond. Unusually short non-covalent F⋯P and O⋯P intramolecular electrostatic contacts also result from the geometrical constraints. The new cage-shaped phosphatriptycene nitrogen-ylides might have interesting reactivity in terms of frustrated Lewis pairs catalysis21 which is subject to further exploration in our labs. The transition metal-free directed electrophilic borylation22 of the triptycene core based on the N-centred directing group approach is also under investigation.
We acknowledge the University of Namur (CERUNA PhD fellowship for S. C.) and the Fond de la Recherche Scientifique FNRS (F.R.S-FNRS) for financial support (Grants T.0012.21 (G. B.) and GEQ UG02222F (J. W.)). Computational resources have been provided by the supercomputing facilities of the Université catholique de Louvain (CISM/UCLouvain) and the Consortium des Équipements de Calcul Intensif en Fédération Wallonie Bruxelles (CÉCI) funded by the F.R.S.-FNRS under convention 2.5020.11 and by the Walloon Region. We thank the PC2 (UNamur) technological platforms for access to all characterization instruments. LH expresses gratitude for the PhD grant support from the China Scholarship Council (CSC, no. 201606670003) and acknowledges the FNRS Mobility Grant for research exchange at the University of Basel. Special appreciation goes to Prof. Olivier Baudoin for hosting and to Dr A. Clemenceau for experimental assistance during this period. RR is a Maître de Recherche of the F.R.S.-FNRS.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS; (b) S. P. Marsden, et al. , Org. Lett., 2008, 10, 2589–2591 CrossRef CAS PubMed; (c) L. Wang, et al. , Synthesis, 2015, 3522–3528 CAS; (d) H. A. Van Kalkeren, et al. , Adv. Synth. Catal., 2012, 354, 1417–1421 CrossRef CAS; (e) H. A. Van Kalkeren, et al. , Eur. J. Org. Chem., 2013, 7059–7066 CrossRef CAS; (f) H. Bel Abed, et al. , Org. Biomol. Chem., 2014, 12, 7159–7166 RSC.
- (a) L. Beaufort, et al. , J. Mol. Catal. A: Chem., 2008, 283, 77–82 CrossRef CAS; (b) O. Alhomaidan, et al. , Organometallics, 2008, 27, 6343–6352 CrossRef CAS; (c) E. Martinez-Arripe, et al. , Organometallics, 2012, 31, 4854–4861 CrossRef CAS; (d) J. García-Álvarez, et al. , J. Organomet. Chem., 2014, 751, 792–808 CrossRef.
- (a) T. Cantat, et al. , Dalton Trans., 2008, 1957–1972 RSC; (b) M. Fustier-Boutignon, et al. , Chem. Rev., 2019, 119, 8555–8700 CrossRef CAS PubMed; (c) K. Dehnicke and F. Weller, Coord. Chem. Rev., 1997, 158, 103 CrossRef CAS; (d) D. W. Stephan, Adv. Organomet. Chem., 2006, 54, 267 CrossRef CAS.
- D. Rozsar, et al. , Angew. Chem., Int. Ed, 2023, 62, e202303391 CrossRef CAS PubMed.
- A. D. Kosal, et al. , Angew. Chem., Int. Ed., 2012, 21, 12036–12040 CrossRef PubMed.
- (a) A. Stute, et al. , Chem. Commun., 2012, 48, 11739–11741 RSC; (b) J. Zhou, et al. , Dalton Trans., 2017, 46, 9334–9338 RSC.
- (a) M. Formica, et al. , Acc. Chem. Res., 2020, 53, 2235–2247 CrossRef CAS PubMed; (b) R. Schwesinger, et al. , Angew. Chem., Int. Ed. Engl., 1993, 32, 1361 CrossRef.
- (a) C. F. Jiang and D. W. Stephan, Dalton Trans., 2013, 42, 630–637 RSC; (b) J. Y. Zhang, et al. , Chin. Chem. Lett., 2018, 29, 1226–1232 CrossRef CAS; (c) S. Döring, et al. , Organometallics, 1998, 17, 2183–2187 CrossRef.
- (a) T. S. Barnard and M. R. Mason, Organometallics, 2001, 20, 206–214 CrossRef CAS; (b) S. Tretiakov, et al. , Angew. Chem., Int. Ed., 2021, 60, 9618–9626 CrossRef CAS PubMed; (c) For a recent review on p-block compounds see: T. J. Hannah and S. S. Chitnis, Chem. Soc. Rev., 2024, 53, 764–792 RSC.
- J. Bedard, et al. , Angew. Chem., Int. Ed., 2022, 61, e202204851 CrossRef CAS PubMed.
- Y.-C. Lin, et al. , Chem. Sci., 2018, 9, 4338–4347 RSC.
- (a) Y. Uchiyama, et al. , J. Org. Chem., 2022, 87, 15899–15913 CrossRef CAS PubMed ; For further applications of phosphatriptycenes in catalysis and coordination chemistry, see: ; (b) Y. Cao, et al. , Organometallics, 2019, 38, 1868 CrossRef CAS.
- (a) L. Hu, et al. , J. Org. Chem., 2019, 84, 11268–11274 CrossRef CAS PubMed; (b) H. Gildenast, et al. , Dalton Trans., 2022, 51, 7828–7837 RSC; (c) D. Rottschäfer, et al. , Inorg. Chem., 2023, 62, 18228 CrossRef PubMed; (d) For the original synthesis of 9-phosphatriptycene, see: C. Jongsma, et al. , Tetrahedron, 1974, 30, 3465–3469 CrossRef CAS.
- (a) H. Kooijman, et al. , Acta Cryst. C, 1695, 1998, 54 Search PubMed; (b) L. R. Falvello, et al. , CSD Commun., 2002 Search PubMed , CCDC 186887; (c) J. Yang, et al. , Chem. Commun., 2016, 52, 12233 RSC; (d) C. Foces-Foces and A. L. Llamas-Saiz, Acta Cryst. C, 1998, 54, 9800013 CrossRef; (e) P. W. Codding and K. A. Kerr, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3785 CrossRef; (f) R. Kumar, et al. , Eur. J. Inorg. Chem., 2018, 1028 CrossRef CAS; (g) A. L. Rheingold, CSD Commun., 2011 Search PubMed , CCDC 856709; (h) H. Hu, et al. , Dalton Trans., 2021, 50, 4772–4777 RSC.
- F. L. Lin, et al. , J. Am. Chem. Soc., 2005, 127, 2686–2695 CrossRef CAS PubMed.
- A similar analysis was made by Dehnicke, See: H. Folkerts, et al. , Angew. Chem., Int. Ed. Engl., 1995, 34, 1362 CrossRef CAS.
- (a) W. Han, et al. , Org. Lett., 2022, 24, 6247–6251 CrossRef CAS PubMed; (b) C. Larré, et al. , Eur. J. Inorg. Chem., 1999, 601–611 CrossRef; (c) J. Yang, et al. , Chem. Commun., 2016, 52, 12233 RSC; (d) Y. Matano, et al. , J. Am. Chem. Soc., 2001, 123, 10954–10965 CrossRef CAS PubMed; (e) E. Böhm, et al. , Zeit. Naturforsch. B, 2014, 43, 138 CrossRef; (f) I. Bar and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 1962–1964 CrossRef.
- T. W. Hudnall and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 10890–10891 CrossRef CAS PubMed.
- (a) N. Fey, et al. , Organometallics, 2008, 27, 1372–1383 CrossRef CAS; (b) J. Jover, et al. , Organometallics, 2012, 31, 5302–5306 CrossRef CAS PubMed; (c) D. J. Durand and N. Fey, Chem. Rev., 2019, 119, 6561–6594 CrossRef CAS PubMed.
- H. Clavier and S. P. Nolan, Chem. Commun, 2010, 46, 841–861 RSC.
- M. G. Guerzoni, et al. , Chem. Catal., 2022, 2, 2865–2875 CrossRef CAS.
- (a) S. Rej and N. Chatani, J. Am. Chem. Soc., 2021, 143, 2920–2929 CrossRef CAS PubMed; (b) S. A. Iqbal, et al. , Chem. Soc. Rev., 2020, 49, 4564–4591 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2313762–2313774 and 2347266–2347270. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01868k |
| ‡ Current address: Xiamen Key Laboratory of Marine Medicinal Natural Product Resources, Xiamen medical college, Xiamen 361023, P. R. China. |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |