
Direct C–H alkylation of 3,4-dihydroquinoxaline-2-ones with N-(acyloxy)phthalimides via radical–radical cross coupling†
Sudhir Kumar
Hota
,
Gulshan
Singh
and
Sandip
Murarka
 *
*
Department of Chemistry, Indian Institute of Technology Jodhpur, Karwar-342037, Rajasthan, India. E-mail: sandipmurarka@iitj.ac.in
First published on 23rd May 2024
Abstract
We present an organophotoredox-catalyzed direct Csp3–H alkylation of 3,4-dihydroquinoxalin-2-ones employing N-(acyloxy)pthalimides to provide corresponding products in good yields. A broad spectrum of NHPI esters (1°, 2°, 3°, and sterically encumbered) participates in the photoinduced alkylation of a variety of 3,4-dihydroquinoxalin-2-ones. In general, mild conditions, broad scope with good functional group tolerance, and scalability are the salient features of this direct alkylation process.
The C–C bond forming reactions have revolutionized organic synthesis, particularly in pharmaceutical industry and materials science. To improve the prospect of clinical success, there is a growing demand in medicinal chemistry to synthesize sp3-enriched drug candidates.1 However, despite substantial progress, Csp3–Csp3 coupling remains a significant challenge and has attracted attention of numerous synthetic chemists.2 In this regard, N-(acyloxy)phthalimides (NHPI esters) that can readily be prepared from feedstock carboxylic acids have emerged as a surrogate of alkyl halides in a variety of radical cross-couplings (RCC) under thermal, photochemical and electrochemical conditions.3 In 2016, the Baran group documented a nickel-catalyzed alkyl–alkyl cross coupling between NHPI esters and alkyl zinc reagents under non-photoinduced conditions (Scheme 1a(i)).4 The last decade has witnessed revolutionary impact of photoinduced synthetic transformations in the advancement of organic synthesis and catalysis.5 Accordingly, several photoinduced approaches involving NHPI esters have been developed for the generation and subsequent Giese-type addition of resulting alkyl radicals on a variety of activated/unactivated alkenes leading to the construction of alkyl–alkyl bonds.3b Nevertheless, examples of Csp3–Csp3 couplings through direct alkylation of Csp3–H bonds with alkyl NHPI esters are scarcely explored.6 Noteworthy examples include, Wang, Xu, and coworkers visible-light-induced Cu-catalyzed Csp3–H alkylation of glycines and peptides, and Shang's photoinduced ammonium iodide-catalyzed C–H alkylation of glycines (Scheme 1a(ii) and (iii)).6a,d Additionally, Cong and Zhang group in their independent reports documented alkylation of benzylic Csp3–H bonds of N-aryl tetrahydroisoquinolines with NHPI esters using either dye-sensitized semiconductor or through electron donor–acceptor (EDA) approach, respectively.6b,c Considering such limited examples, it is of utmost importance to develop novel and sustainable methods enabling Csp3–H functionalizations on a variety of other important substrate classes using NHPI esters as alkyl progenitors.
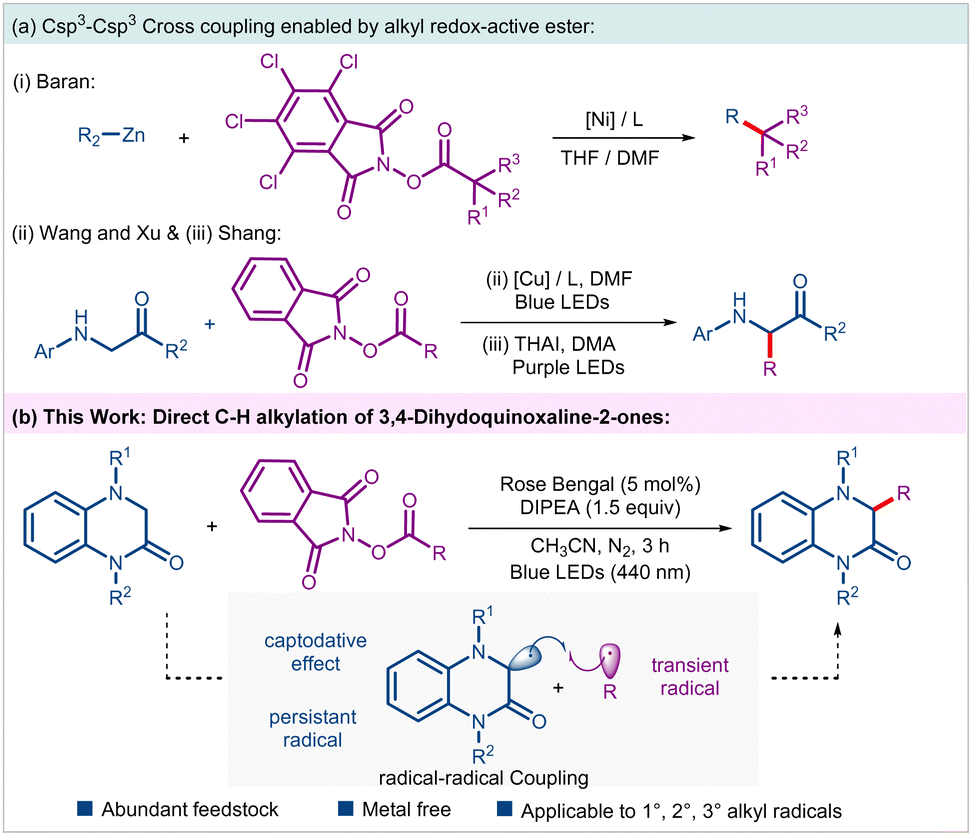 | ||
| Scheme 1 Csp3–Csp3 cross coupling of alkyl N-(acyloxy)phthalimides and proposed work. | ||
3,4-Dihydroquinoxalin-2-ones are a unique class of nitrogen-containing heterocycles that have garnered significant interest from synthetic organic chemists owing to their rich medicinal properties. Compounds embedded with such motifs have shown antiviral, anti-inflammatory, anticancer and other pharmacological properties.7 Accordingly, direct C–H functionalization of this class of compounds enabling tapping of unchartered territory of chemical space have been explored.8 To the best of our knowledge, Csp3–H functionalization of such motifs with an unactivated alkyl group leading to direct Csp3–Csp3 coupling has not been realized till date. These facts inspired us to examine the feasibility of an alkyl–alkyl cross coupling between 3,4-dihydroquinoxalin-2-ones and alkyl NHPI esters under mild photoinduced conditions. In this envisaged process, the transient alkyl radical generated through reductive disintegration of alkyl NHPI esters should undergo radical–radical coupling with the α-amino radical generated from 3,4-dihydroquinoxalin-2-ones under suitable conditions (Scheme 1b). Notably, hetero-selectivity in such diffusion-controlled radical–radical couplings is steered by a kinetic phenomenon called persistent radical effect.9 Nevertheless, such an intriguing direct Csp3–H functionalization strategy allowing alkyl–alkyl coupling would be efficient and rewarding from the perspective of sustainable synthesis, especially if carried out under organophotoredox-catalyzed conditions. Accordingly, as part of our program on the photoinduced synthetic transformations involving NHPI esters,10 we report an organophotoredox-catalyzed alkylation of 3,4-dihydroquinoxalin-2-ones leading to the synthesis of an array of hitherto unexplored alkylated 3,4-dihydroquinoxalin-2-ones in an efficient fashion.
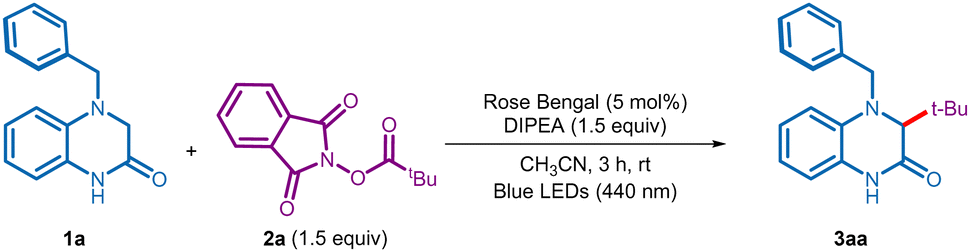
After initial studies, we realized that the reaction was best conducted when a solution of 4-benzyl-3,4-dihydroquinoxalin-2(1H)-one 1a (0.1 mmol, 1 equiv.), NHPI ester 2a (1.5 equiv.), N,N-diisopropylethylamine (DIPEA, 1.5 equiv.), and rose bengal (RB, 5 mol%) in acetonitrile (CH3CN) was irradiated using blue LEDs (440 nm) under nitrogen atmosphere (entry 1, Table 1, see Table S1 in the ESI† for complete optimization). Gratifyingly, under these conditions the reaction was completed in 3 h to provide the desired tert-butylated 3,4-dihydroquinoxalin-2(1H)-one 3aa in 90% yield (entry 1). Replacing rose bengal with other commonly employed photocatalysts, such as eosin Y, and Ru(bpy)3Cl2·6H2O failed to better the yield (entry 2). A quick screening of solvents and bases established CH3CN and DIPEA as the optimal combination (entries 3 and 4). Control experiments indicated the indispensable role of the base, photocatalyst, and visible light irradiation towards the success of this alkyl–alkyl coupling process (entries 5–7).
|
|
||
|---|---|---|
| Entry | Variation from optimized conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol, 1 equiv.), 2a (1.5 equiv.), rose bengal (5 mol%), DIPEA (1.5 equiv.), and CH3CN (1 mL) under nitrogen atmosphere using blue LEDs (440 nm) for 3 h. b Isolated yield. N.D. = not detected. | ||
| 1 | None | 90 |
| 2 | Eosin Y, Ru(bpy)3Cl2·6H2O instead of RB | 66, 85 |
| 3 | THF, CH2Cl2 instead of CH3CN | 71, 61 |
| 4 | TMEDA, Et3N instead of DIPEA | 64, 46 |
| 5 | Without DIPEA | N.D. |
| 6 | Without rose bengal | N.D. |
| 7 | Without irradiation | N.D. |
Having optimized the reaction conditions, we set out to explore the scope of the process. Other than pivaloyl radical, tertiary alkyl radicals derived from 1-methyl-1-cyclohexanecarboxylic acid and 2,2-dimethylbutanoic acid underwent smooth transformation to provide respective products (3ab–3ac) in good yields (80–85%) (Scheme 2). Pleasingly, several secondary alkyl NHPI esters derived from acyclic, such as isopropyl carboxylic acid, as well as cyclic, such as cyclopentyl and cyclohexyl carboxlic acids, 4,4-difluorocyclohexanecarboxylic acid, and N-boc-4-peridinecarboxylic acid successfully engaged in the process to provide corresponding products (3ad–3ah) in moderate to good yields (68–78%). To our delight, more challenging primary alkyl radicals were found to be equally efficient. A palette of primary NHPI esters derived from various carboxylic acids afforded the desired products (3ai–3an) in moderate to excellent yields (60–90%). Notably, primary NHPI esters with terminal alkyne (3am, 62%), as well as derived from α-heteroatom-substituted carboxylic acid (3an, 74%) were compatible under the reaction conditions. A wide range of natural products and pharmaceuticals bearing primary, secondary, and tertiary carboxylic acids, such as stearic acid (3ao, 66%), palmitic acid (3ap, 64%), oleic acid (3aq, 65%), isoxepac (3ar, 72%), ibuprofen (3as, 74%), and gemfibrozil (3at, 82%) were amenable to the C–H alkylation process.
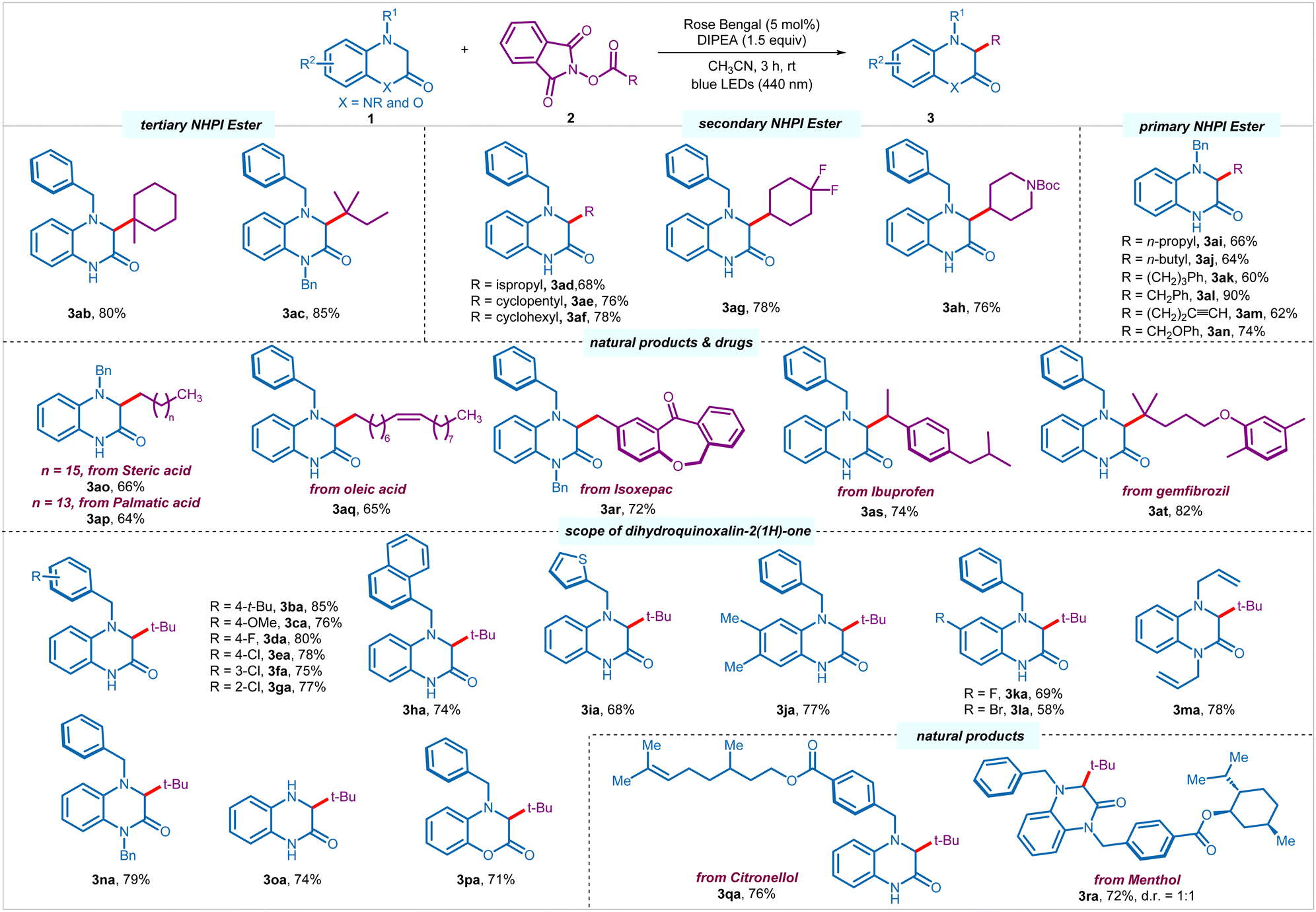 | ||
Scheme 2 Scope of the photoinduced C–H alkylation process.a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1 (0.25 mmol, 1 equiv.), 2 (0.375 mmol, 1.5 equiv.), rose bengal (5 mol%), DIPEA (0.375 mmol, 1.5 equiv.), and CH3CN (2.5 mL) under nitrogen atmosphere using blue LEDs (440 nm) for 3 h. Reaction conditions: 1 (0.25 mmol, 1 equiv.), 2 (0.375 mmol, 1.5 equiv.), rose bengal (5 mol%), DIPEA (0.375 mmol, 1.5 equiv.), and CH3CN (2.5 mL) under nitrogen atmosphere using blue LEDs (440 nm) for 3 h. | ||
Subsequently, we investigated the scope of 3,4-dihydroquinoxalin-2-ones 1 by reacting with alkyl NHPI ester 2a under the optimized conditions (Scheme 2). Several N4-benzylated 3,4-dihydroquinoxalin-2-ones with diverse electron-donating (4-t-Bu, 4-OMe), and electron-withdrawing (4-F, 4-Cl, 3-Cl, 2-Cl) substituents on the phenyl ring of benzyl moiety underwent facile transformation to the desired products (3ba–3ga) in good yields (75–85%), regardless of the position of the substituents. Pleasingly, the aryl functionality at the N4-position could also be replaced with naphthyl (3ha, 74%) and thiophenyl (3ia, 68%) group without affecting the efficacy of the process. Moreover, the core aromatic ring of the heterocycle could also be orchestrated with electron-donating and electron-withdrawing substitution pattern, such as 6.7-di-Me, 6-F, and 6-Br, furnishing desired products (3ja–3la) in good yields (58–77%). To our delight, 1,4-disubstituted (allyl and benzyl) 3,4-dihydroquinoxalin-2-ones were also tolerated to give corresponding products 3ma and 3na in 78% and 79% yields in a respective manner. Notably, unprotected 3,4-dihydroquinoxalin-2(1H)-one with synthetic handle for further modifications was compatible under the reaction conditions to provide 3oa in 74% yield. Interestingly, other heterocycles, such as 4-benzyl-3,4-dihydro-2H-benzo[b][1,4]oxazin-2-one participated in the direct alkylation to provide the corresponding product 3pa in 71% yield. Moreover, 3,4-dihydroquinoxalin-2-ones stitched with various natural products and pharmaceuticals provided desired products (3qa and 3ra) in good yields (76% and 72%).
The scalability and robustness of the method was demonstrated by reacting 1 g of 3,4-dihydroquinoxalin-2-one 1a with NHPI ester 2a under the optimized conditions to afford desired 3aa in 79% yield (see Scheme S1 in the ESI†). To understand the intermediacy of radicals in these transformations, 1a and 2a were reacted in the presence of radical scavengers, such as TEMPO (2,2,6,6-tetramethyl-1-piperidine-1-oxyl) (see Scheme S1 in the ESI†). Expectedly, product formation was not recorded under these conditions, rather, radical trapping adducts 4 and 5 were identified through high-resolution mass spectrometry (HRMS) (Scheme S1 in the ESI†). To further our understanding of the mechanism, we carried out in-depth photophysical investigations. The “light-on-off” experiment in combination with quantum yield (Φ = 0.11) measurements concluded that the desired product 3aa was formed only during the steady irradiation of light and the reaction did not proceed through radical chain mechanism (please see ESI†).11 In the steady-state Stern–Volmer experiments, significant quenching of the fluorescence emission band of rose bengal with 3,4-dihydroquinoxalin-2-one 1a, rather than with 2a or DIPEA was observed (see Scheme S1 in the ESI†). These quenching studies implied that the reaction was initiated through a reductive quenching of the photocatalyst.
Based on the above findings and literature precedence,5f,6a,d,8c,d,12 we propose that the reaction is initiated through photoexcitation of the catalyst to generate an excited state RB*, which upon reductive quenching with 3,4-dihydroquinoxalin-2-ones, such as 1n affords reduced photocatalyst (RB˙−) and radical–cation species A (Scheme 3). The intermediate A upon DIPEA-assisted deprotonation provides persistent α-amino radical B. Notably, in the absence of NHPI ester 2a, formation of dimer 6 was detected, thereby reaffirming the existence of α-amino radical B in the reaction. On the other hand, RB˙− reduces NHPI esters 2a through SET to generate radical–anion C, thereby completing the photocatalytic cycle. C then undergoes disintegration with concomitant elimination of phthalimide anion and carbon dioxide to produce transient alkyl radical D. Finally, radical–radical coupling between B and D results in the formation of cross-coupling product 3na.
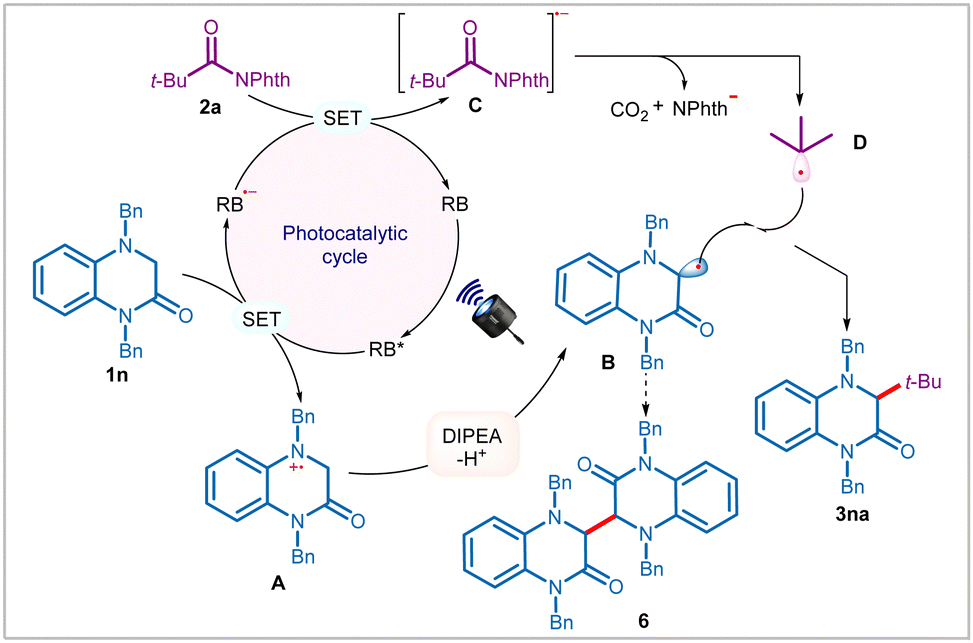 | ||
| Scheme 3 Proposed reaction mechanism. | ||
In summary, we have described a visible light photoredox-catalyzed direct C–H alkylation of 3,4-dihydroquinoxalin-2-ones using alkyl NHPI esters as alkyl radical surrogates. The method exhibits mild conditions, broad scope, and appreciable functional group tolerance. A broad variety of NHPI esters participated in the alkylation of an array of 3,4-dihydroquinoxalin-2-ones to provide corresponding products in good to excellent yields. Detailed mechanistic studies provided explicit insight into the reaction mechanism. Our ongoing research activities focus on developing novel photoinduced synthetic transformation involving redox-active esters enabling tapping of unchartered territory of chemical space.
SM acknowledges SERB [CRG/2022/000470], and CSIR [02(0426)/21/EMR-II] for funding and DST-FIST [SR/FST/CS-II/2019/119(C)] for the HRMS facility at IIT Jodhpur.
Conflicts of interest
“There are no conflicts to declare”.Notes and references
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) F. Lovering, Med. Chem. Commun., 2013, 4, 515–519 RSC.
- (a) M. S. Kharasch, J. K. Hambling and T. P. Rudy, J. Org. Chem., 1959, 24, 303–305 CrossRef CAS; (b) V. D. Parker, L. H. Piette, R. M. Salinger and C. R. Noller, J. Am. Chem. Soc., 1964, 86, 1110–1112 CrossRef CAS; (c) J. K. Kochi and M. Tamura, J. Am. Chem. Soc., 1971, 93, 1485–1487 CrossRef CAS; (d) J. Terao, H. Watanabe, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2002, 124, 4222–4223 CrossRef CAS PubMed; (e) K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2009, 131, 18042–18043 CrossRef CAS PubMed; (f) O. Vechorkin and X. Hu, Angew. Chem., Int. Ed., 2009, 48, 2937–2940 CrossRef CAS PubMed; (g) J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
- (a) S. K. Parida, S. K. Hota, R. Kumar and S. Murarka, Chem. – Asian J., 2021, 16, 879–889 CrossRef CAS PubMed; (b) S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS; (c) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (d) X. Zhu and H. Fu, Chem. Commun., 2021, 57, 9656–9671 RSC; (e) K. Okada, K. Okamoto and M. Oda, J. Am. Chem. Soc., 1988, 110, 8736–8738 CrossRef CAS; (f) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS.
- T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) S. Kumar Hota, D. Jinan, S. Prakash Panda, R. Pan, B. Sahoo and S. Murarka, Asian J. Org. Chem., 2021, 10, 1848–1860 CrossRef CAS; (c) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (e) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (f) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- (a) G.-Z. Wang, M.-C. Fu, B. Zhao and R. Shang, Sci. China: Chem., 2021, 64, 439–444 CrossRef CAS; (b) P. Ma, Y. Liu, L. Chen, X. Zhao, B. Yang and J. Zhang, Org. Chem. Front., 2021, 8, 2473–2479 RSC; (c) L. Ren and H. Cong, Org. Lett., 2018, 20, 3225–3228 CrossRef CAS PubMed; (d) C. Wang, M. Guo, R. Qi, Q. Shang, Q. Liu, S. Wang, L. Zhao, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 15841–15846 CrossRef CAS PubMed.
- (a) Y. Yang, L. Zhao, B. Xu, L. Yang, J. Zhang, H. Zhang and J. Zhou, Bioorg. Chem., 2016, 68, 236–244 CrossRef CAS PubMed; (b) D. J. Hayes, T. M. Mosher and A. J. Greenshaw, Behav. Brain Res., 2009, 197, 323–330 CrossRef CAS PubMed; (c) J. Ren, C. E. Nichols, P. P. Chamberlain, K. L. Weaver, S. A. Short, J. H. Chan, J.-P. Kleim and D. K. Stammers, J. Med. Chem., 2007, 50, 2301–2309 CrossRef CAS PubMed; (d) J. J. Chen, W. Qian, K. Biswas, V. N. Viswanadhan, B. C. Askew, S. Hitchcock, R. W. Hungate, L. Arik and E. Johnson, Bioorg. Med. Chem. Lett., 2008, 18, 4477–4481 CrossRef CAS PubMed.
- (a) J. Rostoll-Berenguer, F. J. Sierra-Molero, G. Blay, J. R. Pedro and C. Vila, Adv. Synth. Catal., 2022, 364, 4054–4060 CrossRef CAS; (b) J. Rostoll-Berenguer, V. García-García, G. Blay, J. R. Pedro and C. Vila, ACS Org. Inorg. Au, 2023, 3, 130–135 CrossRef CAS PubMed; (c) W. Xiong, W.-B. Qin, Y.-S. Zhao, K.-Z. Fu and G.-K. Liu, Org. Chem. Front., 2022, 9, 2141–2148 RSC; (d) J. Rostoll-Berenguer, G. Blay, J. R. Pedro and C. Vila, Org. Lett., 2020, 22, 8012–8017 CrossRef CAS PubMed; (e) J. Rostoll-Berenguer, M. Martín-López, G. Blay, J. R. Pedro and C. Vila, J. Org. Chem., 2022, 87, 9343–9356 CrossRef CAS PubMed.
- (a) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed; (b) L. Zhang, Y. Li, Z. Guo, Y. Li, N. Li, W. Li, C. Zhu and J. Xie, Chin. J. Catal., 2023, 50, 215–221 CrossRef CAS; (c) M. Miao, L.-L. Liao, G.-M. Cao, W.-J. Zhou and D.-G. Yu, Sci. China: Chem., 2019, 62, 1519–1524 CrossRef CAS; (d) X.-Y. Chen, X. Kuang, Y. Wu, J. Zhou and P. Wang, Chin. J. Chem., 2023, 41, 1979–1986 CrossRef CAS; (e) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193–5203 RSC; (f) W. Li, Y. Duan, M. Zhang, J. Cheng and C. Zhu, Chem. Commun., 2016, 52, 7596–7599 RSC; (g) J. Xie, J. Yu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 9416–9421 CrossRef CAS PubMed.
- (a) S. Das, S. K. Parida, T. Mandal, S. K. Hota, L. Roy, S. De Sarkar and S. Murarka, Org. Chem. Front., 2021, 8, 2256–2262 RSC; (b) S. K. Hota, S. P. Panda, S. Das, S. K. Mahapatra, L. Roy, S. De Sarkar and S. Murarka, J. Org. Chem., 2023, 88, 2543–2549 CrossRef CAS PubMed; (c) S. P. Panda, S. K. Hota, R. Dash, L. Roy and S. Murarka, Org. Lett., 2023, 25, 3739–3744 CrossRef CAS PubMed; (d) S. Das, A. Azim, S. K. Hota, S. P. Panda, S. Murarka and S. De Sarkar, Chem. Commun., 2021, 57, 13130–13133 RSC; (e) S. Das, S. K. Parida, T. Mandal, L. Sing, S. De Sarkar and S. Murarka, Chem. – Asian J., 2020, 15, 568–572 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- (a) H. Xue, M. Guo, C. Wang, Y. Shen, R. Qi, Y. Wu, Z. Xu and M. Chang, Org. Chem. Front., 2020, 7, 2426–2431 RSC; (b) W. Ding, L.-Q. Lu, J. Liu, D. Liu, H.-T. Song and W.-J. Xiao, J. Org. Chem., 2016, 81, 7237–7243 CrossRef CAS PubMed; (c) C.-M. Chan, Q. Xing, Y.-C. Chow, S.-F. Hung and W.-Y. Yu, Org. Lett., 2019, 21, 8037–8043 CrossRef CAS PubMed; (d) M. Hari Babu and J. Sim, Eur. J. Org. Chem., 2022, e202200859 CrossRef CAS; (e) J. Rostoll-Berenguer, G. Blay, J. R. Pedro and C. Vila, ChemCatChem, 2023, 15, e202300177 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01837k |
| This journal is © The Royal Society of Chemistry 2024 |