
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative heterometallic catalysts: balancing activity and control in PCL-block-PLA copolymer synthesis†
Maisarah
Abdul Rahman
 ,
Thomas J.
Neal
and
Jennifer A.
Garden
*
,
Thomas J.
Neal
and
Jennifer A.
Garden
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: j.garden@ed.ac.uk
First published on 2nd May 2024
Abstract
Heterometallic cooperativity is gaining momentum in cyclic ester ring-opening polymerisation, yet remains surprisingly underexplored in their block copolymerisations. Here, we report the first homogeneous heterometallic “ate” catalysts for poly(ε-caprolactone)–poly(lactic acid) block copolymers, showcasing the substantial differences in the polymer structures observed upon exchanging Zn for Mg or Ca.
The ring-opening polymerisation (ROP) of cyclic esters produces aliphatic polyesters that are promising alternatives to conventional hydrocarbon-based polymers, yet their applications remain limited by their material properties. For example, poly(lactic acid) (PLA) is bioderived and biodegradable but is also brittle.1 This can be overcome by copolymerising the parent monomer, lactide (LA), with other cyclic esters such as ε-caprolactone (CL), to harness the complementary thermal, physical and mechanical properties of both polymers.2–4 The material properties can be further tuned by altering the copolymer composition, chain lengths and architecture (e.g. block, gradient or random). For example, diblock and triblock copolymers of poly(ε-caprolactone) (PCL)–PLA and PCL–PLA–PCL can give higher elongation at break values than PLA.5 Accordingly, PCL–PLA copolymers have diverse applications in biomedicine,6 compatibilisation7 and packaging production.8 The sequential addition of two (or more) cyclic ester monomers to a metal-based catalyst is a particularly attractive method of producing block copolymers (BCPs), as this is a simple, one-pot, living polymerisation method where some catalysts have delivered high activities and exquisite polymerisation control.9 However, transesterification can disrupt the block structure to give random copolymers, and thus the relative rates of propagation and transesterification are key to controlling the copolymer structures.10
Some of us previously reported a bis-Zn catalyst for cyclic ester ROP (1, Fig. 1), which successfully generated PCL–PLA BCPs via sequential monomer addition, without transesterification disrupting the structure.10 The analogous heterometallic Mg/Zn (2) and Ca/Zn (3) catalysts successfully homopolymerised LA and CL, outperforming homometallic 1 with an overall activity trend 3 > 2 > 1 (Fig. 1).11 These performance enhancements were attributed to the “ate” structure of the heterometallic complexes. Pairing a hard metal (e.g. group 1 and 2) with a softer, more carbophilic metal (e.g. Zn) can result in anionic “ate” activation, with transfer of the anionic ligands to the more carbophilic metal (Fig. 1).12,13 This can simultaneously enhance the Lewis acidity of the group 2 metal (i.e. facilitating monomer coordination), and the nucleophilicity of the Zn-Et/OR unit (enhancing ring-opening and enchainment).14,15 Despite these advantages, the use of heterometallic complexes to prepare BCPs from cyclic esters is surprisingly limited.
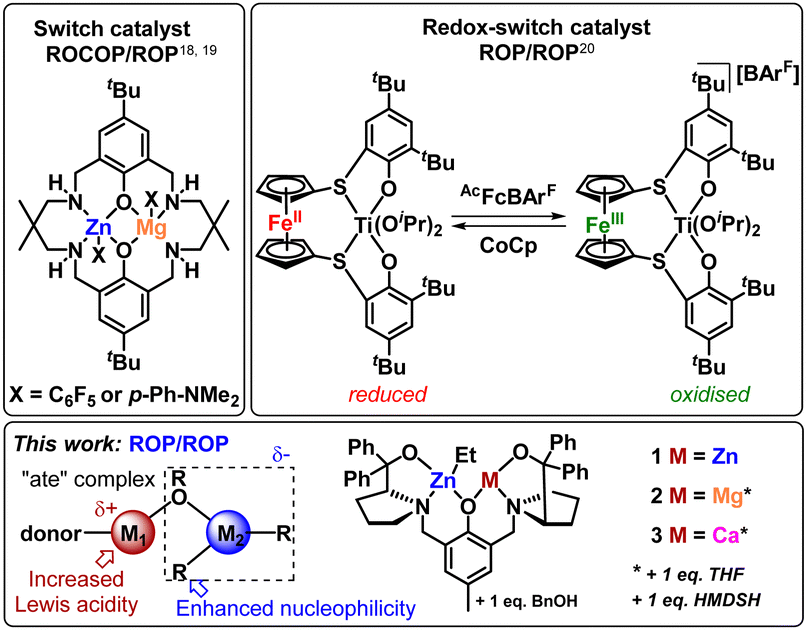 | ||
| Fig. 1 Examples of literature reported heterometallic switch catalysts (top), general structural motifs of “ate” complexes and heterometallic ProPhenol structures tested for cyclic ester block copolymers in this work (bottom). | ||
To the best of our knowledge, the only homogeneous heterometallic catalysts reported for BCPs via cyclic ester ROP are switchable catalysts. These catalysts either switch between cyclic ester ROP and the ring-opening copolymerisation of epoxides with anhydrides or CO2 (Fig. 1, top left),16–19 or use a redox-switchable ferrocene unit to change between LA and CL ROP (Fig. 1, top right).20 The lack of heterometallic catalysts for cyclic ester BCPs is surprising, as several heterometallic catalysts can efficiently homopolymerise multiple cyclic esters.10,21,22 Here, we unlock the potential of heterometallic “ate” catalysts in preparing BCPs from cyclic esters. We also provide insight into the influence of the heterometal on the copolymer structure, revealing which heterometallic catalyst features lead to a trade-off between activity and control.
Complexes 1–3 are efficient catalysts for LA and CL homopolymerisation,10,11 and were selected for this study as the divalent nature of all three metals enables direct comparison between the three catalysts. Therefore, differences in the catalyst activity and copolymer structures can be attributed to the nature of the heterometal instead of differences in the number of co-ligands or initiating units. The synthesis of PCL100–PLA100 diblock copolymers was targeted, with CL as the first monomer because the addition of LA to a living PCL* chain is generally more straightforward than the addition of CL to a PLA* chain (*denotes the active chain end).23–25 All three catalysts gave essentially complete conversion of 100 equiv. CL within 5 minutes, after which 100 equiv. of LA was added to the living PCL* chain to form PCL-b-PLA. Heterometallic 3 was significantly more active than 2 or 1, consuming 95% of rac-LA in 20 min, whereas 2 and 1 required 1.5 h and 2.5 h, respectively. Strikingly, 1 displayed a first-order rate constant for the rac-LA ROP (kobs = 0.052 min−1), whereas 2 and 3 were second-order in monomer (kobs = 0.295 min−1 and 1.17 min−1, respectively) under identical reaction conditions (Fig. S1, ESI†). This differs from the homopolymerisation of rac-LA with 2 and 3, where ROP is initiated by the active catalyst rather than a PCL* chain (refer to ESI† for details).
The formation of PCL-b-PLA copolymers was confirmed by monomodal SEC traces that showed a significant increase in the Mn values upon incorporation of rac-LA as the second block (Fig. S2, ESI†). The dispersities were relatively low for all three catalysts (Table 1, entries 1–3), and were generally narrower for the copolymers than the PCL precursors (Fig. S2, ESI†), indicating that the second step of the copolymerisation (LA ROP) was living in nature.23 The formation of a copolymer was also supported by diffusion order NMR spectroscopy (DOSY NMR, Fig. S3, ESI†), as a single diffusion coefficient was observed for both PCL and PLA resonances. The block structure was further investigated by 1H NMR spectroscopy, which showed that the composition of all copolymers was concordant with the targeted 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 PCL:PLA composition (Table 1). The randomness factor, R, gives insight into whether the copolymer has a well-defined block structure (R = 0) or a purely random structure (R = 1, refer to ESI†).26 Gratifyingly, 1–3 all generated highly defined ‘blocky’ PCL-b-PLA structures, with R values from 0.02–0.04. The average monomer sequence lengths in PCL-b-PLA, lCL and lLA, were also relatively close to the target values (entries 1–3), albeit with 3 delivering somewhat shorter than expected sequence lengths.
:50 PCL:PLA composition (Table 1). The randomness factor, R, gives insight into whether the copolymer has a well-defined block structure (R = 0) or a purely random structure (R = 1, refer to ESI†).26 Gratifyingly, 1–3 all generated highly defined ‘blocky’ PCL-b-PLA structures, with R values from 0.02–0.04. The average monomer sequence lengths in PCL-b-PLA, lCL and lLA, were also relatively close to the target values (entries 1–3), albeit with 3 delivering somewhat shorter than expected sequence lengths.
:100:1:1 ratio of [CL]:[LA]:[cat.]:[BnOH] in toluene solvent
| Cat. | Polymera | M n,obs (kg mol−1) | M n,calc (kg mol−1) | Đ | P i | Composition (PCL:PLA)f |
l CL, lLAf,g | R , | PCL | PL(L)A | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T g (°C) | T m (°C) | T g (°C) | T m (°C) | ||||||||||
| a [CL] = 1 M, [LA] = 0.5 M in toluene, 70 °C (refer to ESI for details). b Determined by SEC analysis using polystyrene standards in THF, values uncorrected to enable comparisons between crude homopolymers and copolymers. c Calculated from monomer conversions, Mn,calc = ((MCL × ([M]/[I]) × CL conv.) + (MLA × ([M]/[I]) × LA conv.)), assuming 1 chain per catalyst. d Determined by decoupled 1H{1H} NMR spectroscopy.31 e Values subject to error due to LA epimerisation with 3 (vide infra).32 f Determined from PCL/PLA 1H NMR integrals for purified copolymer samples (refer to ESI for further details). g l: number average sequence length. h Randomness factor, R: R = 0 (blocky structure), R = 1 (fully random). | |||||||||||||
| 1 | 1 | PCL–PLA | 46.3 | 24.9 | 1.19 | 0.51 | 54:46 |
119, 100 | 0.02 | −62.6 | 56.3 | 25.1 | — |
| 2 | 2 | 35.4 | 25.2 | 1.24 | 0.48 | 50:50 |
100, 99 | 0.02 | −62.2 | 55.8 | 30.4 | — | |
| 3 | 3 | 34.1 | 25.3 | 1.29 | 0.49e | 57:43 |
68, 50 | 0.04 | −60.4 | 55.7 | 29.9 | — | |
| 4 | 1 | PCL-PLLA | 47.8 | 25.3 | 1.27 | 1.00 | 55:45 |
62, 50 | 0.04 | −62.8 | 55.3 | 26.0 | 156.5 |
| 5 | 2 | 42.6 | 24.8 | 1.34 | 1.00 | 52:48 |
54, 50 | 0.04 | −62.2 | 54.7 | — | 163.2 | |
| 6 | 3 | 32.6 | 25.0 | 1.42 | 0.62e | 58:42 |
70, 50 | 0.03 | −59.8 | 55.8 | — | — | |
Overall, these results show that 1–3 are highly efficient catalysts that deliver well-defined PCL-b-PLA copolymers. Notably, catalysts 1–3 did not display any stereocontrol for rac-LA ROP, resulting in atactic PLA blocks in all cases.10,11 Upon substituting rac-LA for L-LA, 1 and 2 generated isotactic PLLA blocks (Table 1, entries 4 and 5), whereas 3 gave an atactic PLA block (entry 6). This is attributed to epimerisation, an issue encountered with some other alkaline earth metal-based catalysts.27 Kricheldorf and co-workers investigated L-LA racemisation with different metal salt initiators and observed that catalysts with higher Brønsted basicity (i.e. Ca vs. Mg) generally gave a greater degree of racemisation due to deprotonation of the methine C–H of L-LA.28 The PCL-b-PLA diblock copolymers synthesised by 1–3 all display similar thermal behaviour, with a single melting temperature (Tm ∼ 56 °C) corresponding to the Tm of PCL homopolymer, but no PLA Tm, reflecting the amorphous state of the atactic PLA block (Table 1 and Table S2, entries 1–3, Fig. S5, ESI†). In all cases, two glass transition temperature (Tg) values were observed for PLA and PCL, indicating that insignificant randomisation occurs in the block copolymer chain.29 The comparable thermal properties of the PCL-b-PLA copolymers gives further evidence that 1–3 all delivered BCP structures. When L-LA was used instead of rac-LA, catalysts 1 and 2 gave polymers with a Tm value of 156–158 °C, due to the crystalline phase of the isotactic PLLA block (Table 1, entries 4 and 5 and Fig. S6, ESI†), whereas no Tm was observed in the copolymers produced using 3. This was attributed to the amorphous nature of the PLA block resulting from L-LA epimerisation (entry 6; Fig. S6, ESI†).
As catalysts 1–3 all displayed high activities and delivered well-controlled diblock copolymer structures, we attempted to access higher-order triblock PCL–PLA–PCL copolymers. The PCL-b-PLA precursor was synthesised using higher monomer concentrations ([CL1] = 1.76 M, [LA] = 0.88 M in toluene) with a subsequent batch of neat CL (CL2) added in situ, as increased concentration improved CL2 conversion in the ‘third block’.30 Intriguingly, homometallic 1 outperformed 2 and 3 in the conversion of CL2 as the third block after 24 h, reaching 73% conversion vs. 42% for 2 and 16% for 3 (Table 2, entries 1, 2 and 4). This is a striking difference from the CL and LA homopolymerisations and the synthesis of PCL-b-PLA diblock copolymers, where the activity trend was 3 > 2 > 1. Moreover, only 1 produced a copolymer with a significantly increased Mn value compared to the PCL-b-PLA precursor (Table S3 and Fig. S7, ESI†).
:[LA]:[CL2]:[cat.]:[BnOH] ratio of 100:100:100:1:1 in toluene solvent
| Cat. | CL2 conv.a (%) | M n (kg mol−1) | Đ | PCL:PLA compositionc |
l CL, lLAc,d | R , | |
|---|---|---|---|---|---|---|---|
| a Determined from 1H NMR integrals for PCL/PLA vs. CL/LA monomer integrals. b Determined by SEC analysis using polystyrene standards in THF, values uncorrected to enable comparisons. c Determined from 1H NMR integrals of purified copolymer samples (see ESI for details). d l: number average sequence length. e Randomness factor, R: R = 0 (blocky structure), R = 1 (fully random). f [CL1] = 1.76 M, [LA] = 0.88 M solution in toluene and CL2 added neat, at 70 °C. g [CL1] = 2 M, [LA] = 1 M solution in toluene and CL2 added neat, at 100 °C. | |||||||
| 1f | 1 | 73 | 56.3 | 1.48 | 64:36 |
9, 5 | 0.31 |
| 2f | 2 | 42 | 31.3 | 1.56 | 53:47 |
9, 8 | 0.23 |
| 3g | 2 | 90 | 31.5 | 1.55 | 67:33 |
4, 2 | 0.76 |
| 4f | 3 | 16 | 28.9 | 1.44 | 63:37 |
21, 13 | 0.13 |
| 5g | 3 | 48 | 22.8 | 1.62 | 63:37 |
7, 4 | 0.38 |
These observations indicate that transesterification is initiated by incorporation of CL into the propagating PCL–PLA* chain (Fig. S8–S11, ESI†). This was confirmed by 1H NMR analysis of the PCL–PLA–PCL copolymer structures. This phenomenon has been observed previously, and is proposed to occur via propagation of a CL monomer followed by transesterification via the reactive PCL-[M] alkoxide bond (Fig. S17, ESI†).33 While the direct comparison of the triblock and diblock structures was somewhat limited by the different copolymer compositions, the triblock copolymers clearly have a less well-defined “blocky” structure than the diblock copolymers, as evidenced by the lower average sequence lengths and higher randomness factors, which increased from R = 0.02–0.04 for the diblock copolymers to R = 0.13–0.76 for the triblock copolymers (Table 2 and Table S3, ESI†).34 The loss of the block structure was attributed to transesterification, as evidenced by notable CL–LA linkage resonances at 4.11 ppm in the 1H NMR spectra (Fig. S4, refer to ESI†). Transesterification appears to outcompete the propagation of CL2 when using heterometallic 2 and 3, with 3 giving the poorest control on the basis of the R and l values as well as DOSY analysis. Specifically, the DOSY spectra gave a single diffusion coefficient for the PCL–PLA–PCL copolymers synthesised using 1 and 2, yet multiple diffusion coefficients for the copolymers prepared using 3. The latter indicates the presence of multiple polymer species in solution due to transesterification, which may be catalysed by the large and Lewis acidic Ca coordinating and activating carbonyl units from the polymer chain towards transesterification (Scheme 1).
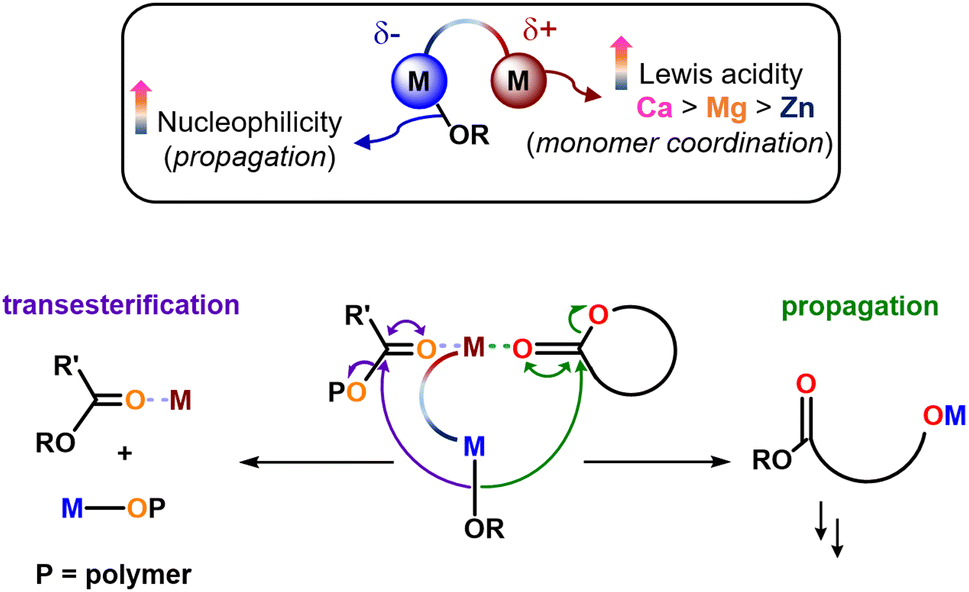 | ||
| Scheme 1 The features of heterometallic “ate” catalysts that enhance propagation also enhance transesterification, with increased metal Lewis acidity (Ca > Mg > Zn) facilitating monomer coordination (propagation) or polymer coordination (transesterification), and enhancing the nucleophilicity of the M-OR unit (both). | ||
To increase the conversion of CL2 using 2 and 3, both the temperature and the monomer concentrations were increased (100 °C, [CL1] = 2 M, [LA] = 1 M, [CL2] = neat, in toluene). This gave higher CL2 conversions, achieving 90% and 48% conversion with 2 and 3, respectively (Table 2, entries 3 and 5). However, this increased conversion comes at the expense of increased transesterification and randomisation of the polymer microstructure, as confirmed by the increased R factors and shorter average sequence lengths (e.g. Table S3, ESI† entry 4, R increases from 0.02 for PCL-b-PLA to 0.76 for PCL–PLA–PCL). Additionally, despite 90% conversion of the third monomer, the Mn value increased only slightly (from 31.0 kg mol−1 for PCL-b-PLA to 31.5 kg mol−1 PCL–PLA–PCL, Fig. S10, ESI†), indicating that intramolecular transesterification occurs forming lower molar mass species (Fig. S8–S11, ESI†).35 The experimental data suggests that with 2 and 3, higher CL2 conversion is generally linked to increased transesterification (Table 2, entries 2 vs. 3 and 4 vs. 5), which makes it challenging to draw direct comparisons between the different catalysts. Yet 3 gives very short average sequence lengths even at just 48% conversion, which are lower than the average sequence lengths observed with catalyst 2 at a similar, albeit slightly lower conversion (entries 2 and 5). Perhaps more significant are the shorter Mn values and the multiple DOSY diffusion coefficients observed with 3 (e.g. entry 5, Fig. S11 and S12, ESI†), which indicate a greater loss of control i.e. more transesterification occurring with 3 than for 2. For example, with 3, the Mn value decreased from 26.4 kg mol−1 to 22.8 kg mol−1 upon addition of CL2, which correlated with a visual decrease in the polymerisation viscosity upon addition of CL2. Taken together, this data suggests that the different catalysts give different degrees of transesterification upon addition of CL to the PCL–PLA* chain, in the order of 3 > 2 > 1, highlighting the influence of the (hetero)metal over the structure of PCL–PLA–PCL copolymers.
Notably, the transesterification trend of 3 > 2 > 1 upon addition of CL2 matches the catalyst propagation rate in the homopolymerisation of CL or LA and the synthesis of PCL–PLA diblock copolymers. Addition of CL2 converts the metal–PLA* alkoxide bond into a metal–PCL* alkoxide bond, and transesterification significantly outcompetes propagation for both 2 and 3, resulting in low conversions of CL2 as the third monomer. This shows that heterometallic catalysts can deliver activity enhancements in ROP but this needs to be carefully balanced, as the catalyst features that lead to enhanced activities can also enhance transesterification side-reactions (Scheme 1 and Fig. S17, ESI†). Specifically, both Lewis acid coordination of a Lewis donor monomer and nucleophilic attack/ring-opening are key steps in cyclic ester ROP.14 Notably, Lewis acid catalysed transesterification also involves the Lewis activation of a carbonyl unit and nucleophilic attack. The nature of the (hetero)metals is thus key in controlling the competition between propagation and transesterification.
To the best of our knowledge, this study represents the first report of a homogeneous, heterometallic “ate” catalyst for the synthesis of well-controlled PCL-b-PLA diblock copolymers. The heterometallic Ca/Zn and Mg/Zn complexes outperform the homometallic bis-Zn analogue in terms of activity, showcasing that heterometallic cooperativity enhancements can be translated from PLA and PCL homopolymers to diblock copolymers. In contrast, investigations into PCL–PLA–PCL triblock copolymers revealed that addition of CL2 as the third monomer triggered transesterification, with heterometallic 2 and 3 giving more random polymer structures than homometallic 1. The enhanced activity of heterometallic complexes in cyclic ester ROP has previously been ascribed to the formation of “ate” structures, which simultaneously enhances the Lewis acidity of one metal (e.g. Mg or Ca) to aid monomer coordination, and the nucleophilicity of another (e.g. Zn–OR bond) to enhance nucleophilic attack and ring-opening.11 While these two catalyst features facilitate ROP, they can also facilitate transesterification through a Lewis acid activation mechanism. Therefore, a careful choice of metal combinations in heterometallic complexes is imperative to harness both high activity and good control in the synthesis of well-defined PCL–P(L)LA BCPs. These studies pave the way for future investigations into harnessing heterometallic cooperativity for the efficient and controlled synthesis of block copolyesters, and other cyclic ester combinations are currently under investigation in our laboratory.
We gratefully acknowledge MARA Malaysia (M. A. R.), the UKRI Future Leaders Fellowship (J. A. G. and T. N. MR\T042710\1), British Ramsay Memorial Trust (J. A. G.), L’Oréal-UNESCO for Women in Science UK & Ireland Fellowship (J. A. G.) and British Royal Society (J. A. G. RSG\R1\180101) for funding, as well as Dr Weronika Gruszka for preliminary investigations.
Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Li, Y. Lin, M. Liu, L. Meng and C. Li, J. Appl. Polym. Sci., 2023, 140, e53477 CrossRef CAS.
- C. Jacobs, P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 1991, 24, 3027–3034 CrossRef CAS.
- I. D’Auria, M. C. D’Alterio, C. Tedesco and C. Pellecchia, RSC Adv., 2019, 9, 32771–32779 RSC.
- F. Della Monica, E. Luciano, A. Buonerba, A. Grassi, S. Milione and C. Capacchione, RSC Adv., 2014, 4, 51262–51267 RSC.
- C. X. Song and X. D. Feng, Macromolecules, 1984, 17, 2764–2767 CrossRef CAS.
- B. G. Amsden, M. Y. Tse, N. D. Turner, D. K. Knight and S. C. Pang, Biomacromolecules, 2006, 7, 365–372 CrossRef CAS PubMed.
- A. R. Kakroodi, Y. Kazemi, D. Rodrigue and C. B. Park, Chem. Eng. J., 2018, 351, 976–984 CrossRef CAS.
- D. V. Plackett, V. K. Holm, P. Johansen, S. Ndoni, P. V. Nielsen, T. Sipilainen-Malm, A. Södergård and S. Verstichel, Packag. Technol. Sci., 2006, 19, 1–24 CrossRef CAS.
- C. Diaz and P. Mehrkhodavandi, Polym. Chem., 2021, 12, 783–806 RSC.
- W. Gruszka, L. C. Walker, M. P. Shaver and J. A. Garden, Macromolecules, 2020, 53, 4294–4302 CrossRef CAS.
- W. Gruszka, H. Sha, A. Buchard and J. A. Garden, Catal. Sci. Technol., 2022, 12, 1070–1079 RSC.
- W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 1–13 CrossRef PubMed.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- E. Fazekas, P. A. Lowy, M. Abdul Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- E. Fazekas, P. A. Lowy, M. Abdul Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2023, 52, 1157 RSC.
- Y. Huang, C. Hu, X. Pang, Y. Zhou, R. Duan, Z. Sun and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202202660 CrossRef CAS PubMed.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed.
- J. A. Garden, A. J. P. White and C. K. Williams, Dalton Trans., 2017, 46, 2532–2541 RSC.
- W. Gruszka, A. Lykkeberg, G. S. Nichol, M. P. Shaver, A. Buchard and J. A. Garden, Chem. Sci., 2020, 11, 11785–11790 RSC.
- T. Rosen, I. Goldberg, W. Navarra, V. Venditto and M. Kol, Angew. Chem., Int. Ed., 2018, 57, 7191–7195 CrossRef CAS PubMed.
- A. Pilone, M. Lamberti, M. Mazzeo, S. Milione and C. Pellecchia, Dalton Trans., 2013, 42, 13036 RSC.
- J. Bai, X. Xiao, Y. Zhang, J. Chao and X. Chen, Dalton Trans., 2017, 46, 9846–9858 RSC.
- J. Fernández, A. Etxeberria and J.-R. Sarasua, J. Mech. Behav. Biomed. Mater., 2012, 9, 100–112 CrossRef PubMed.
- R. Platel, L. Hodgson and C. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- H. Kricheldorf and A. Serra, Polym. Bull., 1985, 14, 497–502 CrossRef CAS.
- P. Vanhoorne, P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 1992, 25, 37–44 CrossRef CAS.
- B. O’Shaughnessy, Macromolecules, 1994, 27, 3875–3884 CrossRef.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- T. J. Neal, E. D. Neal, J. Cumby and J. A. Garden, Polym. Chem., 2024, 15, 1704–1713 RSC.
- Y. Baimark and R. Molloy, Polym. Adv. Technol., 2005, 16, 332–337 CrossRef CAS.
- S. Penczek, A. Duda and R. Szymanski, Macromol. Symp., 1998, 132, 441–449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR (1H, 13C and DOSY) spectra and DSC data. See DOI: https://doi.org/10.1039/d4cc01664e |
| This journal is © The Royal Society of Chemistry 2024 |