
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lewis base adducts of NpCl4†
Lauren M.
Lopez‡
 ,
Madeleine C.
Uible‡
,
Matthias
Zeller
and
Suzanne C.
Bart
*
,
Madeleine C.
Uible‡
,
Matthias
Zeller
and
Suzanne C.
Bart
*
H.C. Brown Laboratory of Chemistry, Department of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA. E-mail: sbart@purdue.edu
First published on 3rd May 2024
Abstract
Np(IV) Lewis base adducts were prepared by ligand substitution of NpCl4(DME)2. Using acetonitrile and pyridine, NpCl4(MeCN)4 (1) and NpCl4(pyr)4 (2) were isolated, respectively. Addition of t-butylbipyridine and triphenylphosphine oxide generated the respective Lewis base adducts, NpCl4(tBuBipy)2 (3) and NpCl4(OPPh3)2 (4). All species were fully characterized using spectroscopic and structural analyses.
A major obstacle to the exploration of the fundamental chemistry of the actinides, and more specifically transuranium elements, is the lack of available starting materials.1,2 Developing non-aqueous routes to halides that are soluble in a variety of organic solvents is important because these are not commercially available. Using the available transuranium oxides, non-aqueous halides are generated by digestion in acids followed by treatment with drying agents.3,4 In the case of neptunium, Gaunt and co-workers first reported the synthesis of NpCl4(DME)2 (DME = dimethoxyethane),5 a convenient material used for further synthetic efforts. The synthesis of NpCl4(DME)2, however, requires the use of TMS-Cl, which makes N-based adducts difficult to produce from the original synthesis. The identity of NpCl4(DME)2 is well-established in DME, and it's ligand substitution chemistry has been explored in THF, generating NpCl4(THF)3.6 Its ligand substitution in other solvents is currently unknown.
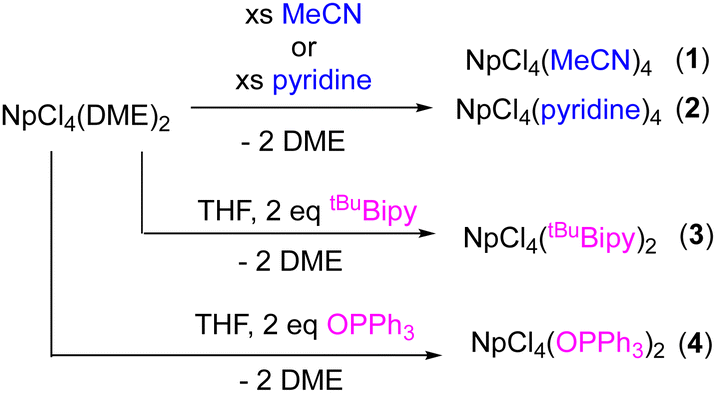
We recently published the synthesis of two Np(III) halides, NpI3(THF)4 and NpBr3(THF)4, which were isolated as THF adducts from neptunia (NpO2), in good yields.3 These materials were straightforward to synthesize and could be used as starting materials for non-aqueous Np(III) synthetic pathways. We were also interested in determining the identity of NpCl4(DME)2 when dissolved in organic solvents commonly used for syntheses of transuranium compounds. Herein, we report the preparation of two new Lewis base solvent adducts of neptunium(IV) chloride, acetonitrile (MeCN) and pyridine (pyr). It was found that the DME was readily replaced by these Lewis basic solvents. To further demonstrate the lability of DME in current Np(IV) starting material, two new compounds, NpCl4(tBuBipy)2 and NpCl4(OPPh3)2, were also synthesized. All four novel products were characterized by 1H NMR spectroscopy, electronic absorption spectroscopy, and X-ray crystallography and then compared to their respective uranium analogues.
The ligand substitution study commenced with the dissolution of pink NpCl4(DME)2 in either MeCN or pyridine (Scheme 1). Each solution was stirred for ∼16 hours, followed by removal of volatiles in vacuo. The acetonitrile product (1) turned very pale pink, whereas the pyridine (2) was a yellow-brown powder. Both materials were isolated in high yields (95% and 93%, respectively). Solid-state infrared spectroscopy of 1 shows absorptions at 2324, 2280 cm−1 assignable to the acetonitrile ligands, which are higher energy than those we observed for free acetonitrile (2293, 2254 cm−1). Under the same conditions, the uranium analogue, UCl4(MeCN)4, which has only previously been crystallographically characterized,7,8 has absorptions at 2307, 2280 cm−1 as well. The strengthening of the N–C triple bond is typical for actinide(IV) nitrile compounds. Neu and co-workers report a uranium(IV) benzonitrile species, UI4(NCPh)4, with a square antiprismatic uranium ion,9 where the absorption for the nitriles appears at 2254 cm−1 as recorded by infrared spectroscopy. This is shifted from that of free benzonitrile (2228 cm−1), and is reported to occur from coordination to the highly electropositive uranium(IV) ion that polarizes the triple bond.9 Similar conclusions can be drawn about 1 and UCl4(MeCN)4.
 | ||
| Scheme 1 Synthesis of 1–4. | ||
Analyses by 1H NMR spectroscopy proved difficult for these adducts due to solubility, fluxionality, and shifting from paramagnetic neptunium ions. The 1H NMR spectrum of 1 (chloroform-d, 25 °C) shows a broad resonance at 1.89 ppm assignable to the methyl groups of the acetonitrile ligands (Fig. S1, ESI†). The extreme broadness noted for this resonance is likely due to a combination of the paramagnetic Np(IV), f3 center and fluxionality of the acetonitrile ligands on the NMR timescale. For comparison, in our hands, the 1H NMR spectrum of UCl4(MeCN)4 (benzene-d6, 25 °C) shows a single resonance at 0.42 ppm assignable to the methyl groups of the acetonitrile ligands coordinated to the uranium(IV), f2 center (Fig. S2, ESI†).
For 2, the 1H NMR spectrum could not be acquired in chloroform-d or any other common NMR solvents due to solubility issues. While 2 is soluble in pyridine-d5 (25 °C), the 1H NMR spectrum did not provide useful structural information due to the pyridine/pyridine-d5 exchange (Fig. S3, ESI†). However, the 1H NMR spectrum of UCl4(pyr)4 was acquired in benzene-d6 (25 °C) in our laboratory and showed three very broad resonances assignable to exchanging pyridine ligands (−8.93, 0.14, and 2.76 ppm) (Fig. S4, ESI†).
Slow cooling of NpCl4(DME)2 in acetonitrile or pyridine from 100 °C produced single crystals suitable for X-ray diffraction of 1 and 2. Refinement of these data revealed 1 as NpCl4(MeCN)4 and 2 as NpCl4(pyr)4 (Fig. 1 and Table 1). Both derivatives have 8-coordinate neptunium centers in distorted dodecahedral geometries, featuring four chlorides and four solvent molecules. The uranium analogue of 1, UCl4(MeCN)4, is also reported to be dodecahedral,7,8 whereas the actinide analogues of 2, UCl4(pyr)410 and ThCl4(pyr)4, were not described.11 The Np–N distances in 1 are 2-fold symmetric, with values of 2.5526(17) and 2.5716(16) Å, whereas those for 2 also with 2-fold symmetry are longer at 2.6857(14) and 2.6877(14) Å. This variation in distances is likely due to sterics, as the acetonitrile ligands in 1 are cylindrical whereas the pyridine ligands in 2 are planar and pack less efficiently. These distances are on par with those reported for Np(OtBu)4(py)2, where the Np–N distances are 2.661(3) and 2.667(3) Å.12
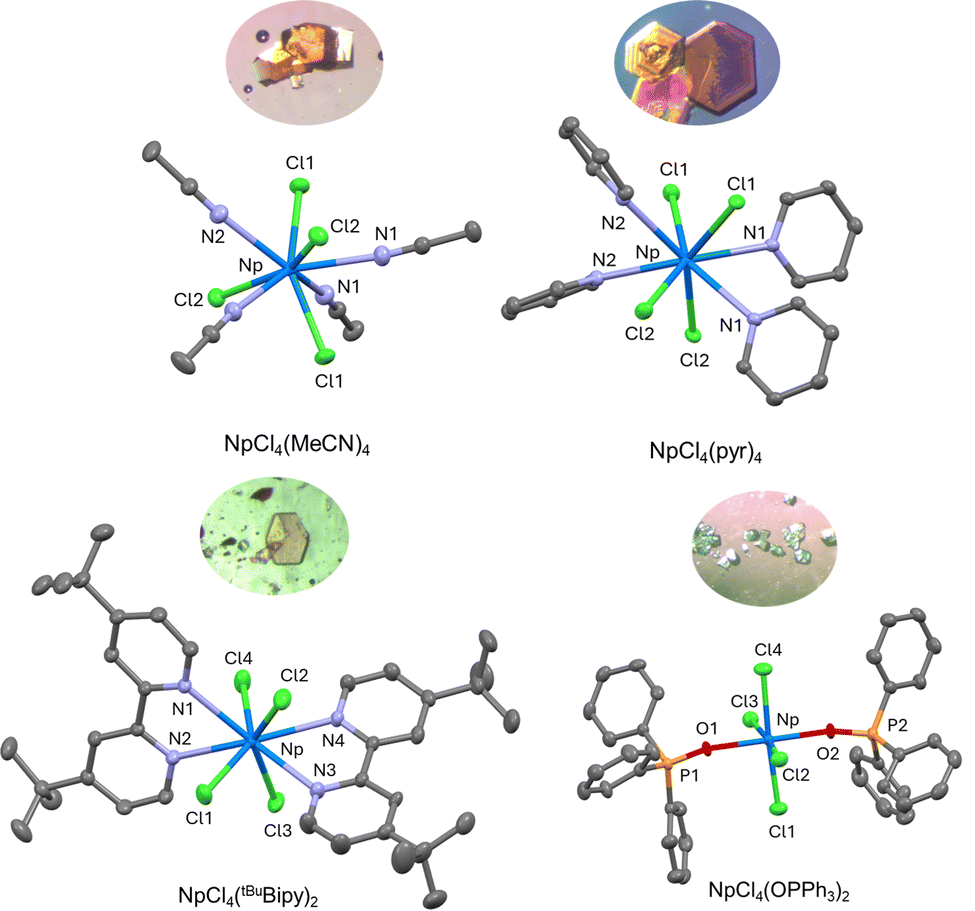 | ||
| Fig. 1 Molecular structures and insets of their crystals of 1 (top-left), 2 (top-right), 3 (bottom-left), and 4 (bottom-right) shown at 50% probability ellipsoids. Hydrogen atoms, co-crystallized solvent molecules, and disorder have been omitted for clarity. | ||
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Np–Cl | 2.6159(4) 2.6067(4) | 2.6269(4) 2.6188(4) | 2.6264(9) | 2.5732(17) |
| 2.6031(9) | 2.6093(15) | |||
| 2.6271(8) | 2.6113(15) | |||
| 2.6282(9) | 2.6148(17) | |||
| Np–N | 2.5526(17) 2.5716(16) | 2.6857(14) 2.6877(14) | 2.628(3) | |
| 2.610(3) | ||||
| 2.650(3) | ||||
| 2.637(3) | ||||
| Np–O | 2.227(4) | |||
| 2.233(4) | ||||
The Np–Cl distances of 1 and 2 track similarly to the Np–N bonds, where those in 1 have values of 2.6159(4) and 2.6067(4) Å, and those in 2 are slightly longer at 2.6269(4) and 2.6188(4) Å. This is again consistent with 2 being more sterically crowded, causing the chloride ligands to have slightly weaker bonds. These agree with those reported for NpCl4(DME)2, which have Np–Cl distances ranging from 2.5878(9) to 2.6224(9) Å,5 and for NpCl4(THF)3 that range from 2.568(2) to 2.607(2) Å.6 The Np–Cl distances in Cp*2NpCl2 are slightly shorter (2.583(6) Å) due to the coordinatively unsaturated neptunium ion in this case.13
Following the characterization of the solvent adducts 1 and 2, ligand substitution chemistry with 4,4′-di-tert-butyl-2,2′-bipyridine (tBuBipy) was targeted, as a neptunium(IV) center has not yet been explored with this popular ligand. To synthesize this, a solution of two equiv. of tBuBipy in THF was prepared and added dropwise to a THF solution of NpCl4(DME)2 (Scheme 1), resulting in an immediate color change from light pink to bright pink. The reaction was then filtered, and volatiles were removed in vacuo. The product, NpCl4(tBuBipy)2 (3), was isolated in good yield (81%). The 1H NMR spectrum of 3 (chloroform-d or benzene-d6 25 °C) is paramagnetically broadened and shifted, as expected for a Np(IV) center, but was not useful for structural assignment. This is in analogy to the NMR data collected by Boncella and coworkers for UCl4(tBuBipy)2, which features a U(IV), f2 center.14
Single crystals of 3 suitable for X-ray diffraction were grown from a concentrated solution of THF layered with pentane at −35 °C (Fig. 1 and Table 1). Refinement of the data obtained showed an eight-coordinate neptunium center with four chlorides and two tBuBipy ligands in an approximate dodecahedral geometry. This is unlike UCl4(tBuBipy)2, which is reported to be square antiprismatic.14 The Np–N distances for 3 of 2.628(3), 2.610(3), 2.650(3), and 2.637(3) Å are shorter than for 2, likely a result of the smaller steric effect of the bipyridine ligand as compared to the pyridine ligand since the bipyridine ligands are chelators and have restricted motion. The Np–Cl bonds in 3, which have distances of 2.6264(9), 2.6031(9), 2.6271(8), and 2.6282(9) Å, do not show significant deviation from those of 1 or 2, indicating that these ligands are less sensitive to steric changes. To our knowledge, this is the first example of a neptunium(IV) bipyridine compound reported crystallographically.
Triphenylphosphine oxide (OPPh3) was also explored for its ligand substitution chemistry with NpCl4(DME)2. This reaction was previously examined by Gaunt and coworkers, but no tractable products were ever characterized. While a neptunium–triphenylphosphine adduct was not isolated or crystallographically characterized, this group did report a white solid that precipitated from solution when OPPh3 was added to a solution of NpCl4(DME)2. In our hands, when two equivalents of OPPh3 were dissolved in THF and added dropwise to a pink solution of NpCl4(DME)2, a colour change to light blue was observed. The solution remained homogeneous, with no white precipitate noted. Removal of the volatiles in vacuo produced a light blue residue (4). The 1H NMR spectrum of 4 (chloroform-d, 25 °C) was paramagnetically broadened and shifted, as expected for a Np(IV) center. While it was difficult to garner structural information, it could be noted that there was a major and minor product present, consistent with formation of two different isomers. This was also supported by 31P NMR spectroscopy (chloroform-d, 25 °C), which showed the presence of a large resonance at 124 ppm with a small shoulder at 102 ppm, indicating two inequivalent but similar phosphorus environments. Infrared spectroscopy of 4 (KBr pellet) showed an absorption around 1121 cm−1, assignable to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P bond stretch (Fig. S16, ESI†).15
P bond stretch (Fig. S16, ESI†).15
Cooling the THF solution to –35 °C, caused light blue crystals suitable for X-ray diffraction to precipitate. Analysis and refinement of the data showed a six-coordinate, octahedral complex assigned as NpCl4(OPPh3)2 (4) (Fig. 1 and Table 1). The two OPPh3 ligands are trans with respect to each other, and the four chlorides are found in a square plane. This arrangement is in contrast to PuCl4(OPPh3)2,16 which is octahedral but found as the cis isomer. The uranium analogue, UCl4(OPPh3)2, has been crystallized in both the cis17 and trans18 forms previously. Thus, the two isomers noted in the solution NMR data were likely the cis and trans forms, but only the trans was noted by X-ray crystallography. The thorium congener, ThCl4(OPPh3)3, can accommodate an extra OPPh3 ligand, likely because of the increased ionic radius of Th(IV) (0.94 Å) as compared to U(IV) (0.89 Å) and Pu(IV) (0.86 Å).16 The Np–O distances of 2.227(4) and 2.233(4) Å are similar to those found in NpCl4(DME)2, which range from 2.520(2) to 2.579(2) Å.5 This slightly shorter distance in 4 is likely due to less sterically hindered Np center, allowing closer approach of the OPPh3 ligands. The P–O distances in 4 were found to be 1.528(4) and 1.523(4) Å, which is on par with those for PuCl4(OPPh3)2 (1.520(4) Å)16 and cis-UCl4(OPPh3)2 (1.524(7) Å).17 Notably, the Np–O–P angles deviate from linearity in 4, with values of 153.6(3)° and 166.6(3)°, similar to those for PuCl4(OPPh3)2 of 163.6(2)° and cis-UCl4(OPPh3)2 of 165.1(5)°.
The electronic structures of 1–4 were interrogated using electronic absorption spectroscopy. Data for these compounds were collected at ambient temperature in the UV-Vis and NIR regions and compared to their uranium counterparts. Due to variation in solubility, data were collected in different solvents along with their uranium analogues. The spectra for 1 and UCl4(MeCN)4 were acquired in both acetonitrile and dichloromethane (Fig. S7–S9, ESI†), whereas those for 2 and UCl4(pyr)4 were collected in pyridine (Fig. 2). The spectrum of 1 in acetonitrile showed strong absorbances at 305 nm (ε = 1700 M−1 cm−1) and 360 nm (ε = 1200 M−1 cm−1), compared to 303 nm (ε = 2600 M−1 cm−1) for the uranium analogue (Fig. S8, ESI†). Both 1 and UCl4(MeCN)4 also had weak absorptions throughout their NIR regions, consistent with either Laporte forbidden 5f–5f transitions or 5f–6d transitions.19 Whereas, in dichloromethane, both 1 and UCl4(MeCN)4 were poorly soluble, generating very broad spectra (Fig. S9 and S10, ESI†). For the pyridine adducts, the neptunium and uranium compounds have similar spectroscopic features. Both show absorptions around 300 nm (U: ε = 2100 M−1 cm−1; Np: ε = 2500 M−1 cm−1), assignable to those for the pyridine ligands, with an additional shoulder at 388 nm (ε = 830 M−1 cm−1) for 2. The Np(IV), f3 and U(IV), f2 centers also show analogous features in the near infrared regions; both of these ions show weak but very sharp absorptions throughout this area, signature for their tetravalent oxidation states and assignable to f–f transitions (Fig. 2).
 | ||
| Fig. 2 Electronic absorption spectra of 2 and UCl4(pyr)4 shown in the UV-Vis and NIR regions at ambient temperature in pyridine. | ||
Similarly to 2, the electronic absorption spectrum for 3, recorded in dichloromethane, shows intense absorptions throughout the UV-Vis and NIR regions signature for the bipyridine ligands (Fig. S12, ESI†). Compound 3 has absorptions at 493 nm (ε = 200 M−1 cm−1) and 530 nm (ε = 240 M−1 cm−1), likely responsible for the observed pink color. This is in comparison to the uranium derivative, UCl4(tBuBipy)2, which has an absorption in the visible at 670 nm (ε = 100 M−1 cm−1), responsible for the observed green color of the complex.20 Additionally, weak but sharp absorptions are also present throughout the NIR regions assignable to the Np(IV), f3 ion. A broad absorption appears in the NIR for the uranium analogue at 1145 nm (ε = 50 M−1 cm−1).
The spectrum for 4, also acquired in dichloromethane, shows a strong absorption in the visible region at 365 nm (ε = 1200 M−1 cm−1) (Fig. 3). Additionally, sharp peaks with low molar absorptivity (ε < 50 M−1 cm−1) can be observed throughout the NIR region of the spectrum (Fig. S13, ESI†). The uranium counterpart lacks this absorption at 365 nm but shows sharp absorptions with low molar absorptivity (ε < 20 M−1 cm−1) in the NIR region. Notably, the uranium analogue displays an absorption at 1930 nm (ε = 30 M−1 cm−1) that is absent in the neptunium congener, demonstrating the differences in electronic structure between NpCl4(OPPh3)2 and UCl4(OPPh3)2.
 | ||
| Fig. 3 Electronic absorption spectra for 3 and 4 recorded in the visible region at ambient temperature in dichloromethane. | ||
In summary, four new neptunium species, compounds 1–4, have been synthesized by ligand substitution of NpCl4(DME)2 and fully characterized using spectroscopic and structural methods. These species have also been compared to their uranium analogues, which are now fully characterized as well. Characterization by NMR spectroscopy highlights the dynamic ligand substitution that occurs when NpCl4(DME)2 is dissolved in pyridine and acetonitrile. DME ligands are also easily replaced by 4,4′-ditertbutyl-2,2′-bipyridine and triphenylphosphine oxide. Compounds 1–3 consist of tetravalent neptunium ions coordinated by Lewis bases in eight-coordinate geometries, whereas 4 is six-coordinate and octahedral. Electronic absorption spectroscopic studies performed in a variety of solvents are all consistent with the Np(IV), f3 electronic structures of these ions. Additionally, the first neptunium(IV) bipyridine and triphenylphosphine oxide compounds have been characterized crystallographically.
Given that the current, most prevalent starting material for non-aqueous neptunium chemistry is NpCl4(DME)2, and 1–4 were made using this DME adduct, our studies give insight into the ligand substitution chemistry that occurs at the Np(IV) ion. These studies support that the DME ligand is easily replaced by stronger ligands. Spectroscopic data confirm that the newly coordinated acetonitrile and pyridine ligands are fluxional in solution on the NMR timescale, but were fully coordinated in the solid state by crystallographic studies. For the triphenylphosphine oxide ligand, two sets of broad resonances are noted in solution supporting fluxional behavior, whereas only one isomer is observed via X-ray crystallography. The chelation of the bipyridine ligand prevents any type of dynamic behavior, indicating this ligand would be a good spectator, but won’t be easily substituted. Additional studies will be aimed at substitution chemistry in these solvents to generate organometallic neptunium derivatives.
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry Program under Award Number DE-SC0008479. We also acknowledge Mr Nathan Lin for assistance with X-ray crystallography.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed.
- P. L. Arnold, M. S. Dutkiewicz and O. Walter, Chem. Rev., 2017, 117, 11460–11475 CrossRef CAS PubMed.
- M. A. Whitefoot, D. Perales, M. Zeller and S. C. Bart, Chem. – Eur. J., 2021, 27, 18054–18057 CrossRef CAS PubMed.
- S. S. Galley, J. M. Sperling, C. J. Windorff, M. Zeller, T. E. Albrecht-Schmitt and S. C. Bart, Organometallics, 2019, 38, 606–609 CrossRef CAS.
- S. D. Reilly, J. L. Brown, B. L. Scott and A. J. Gaunt, Dalton Trans., 2014, 43, 1498–1501 RSC.
- S. A. Pattenaude, N. H. Anderson, S. C. Bart, A. J. Gaunt and B. L. Scott, Chem. Commun., 2018, 54, 6113–6116 RSC.
- F. A. Cotton, D. O. Marler and W. Schwotzer, Acta Cryst. C, 1984, 40, 1186–1188 CrossRef.
- G. Van den Bossche, J. Rebizant, M. R. Spirlet and J. Goffart, Acta Cryst. C, 1986, 42, 1478–1480 CrossRef.
- A. E. Enriquez, B. L. Scott and M. P. Neu, Inorg. Chem., 2005, 44, 7403–7413 CrossRef CAS PubMed.
- J. C. Berthet, P. Thuery and M. Ephritikhine, Private Communication, Cambridge Crystallographic Database, 2013, DOI:10.5517/cc11647m.
- W. Wu, D. Rehe, P. Hrobárik, A. Y. Kornienko, T. J. Emge and J. G. Brennan, Inorg. Chem., 2018, 57, 14821–14833 CrossRef CAS PubMed.
- D. Grödler, J. M. Sperling, B. M. Rotermund, B. Scheibe, N. B. Beck, S. Mathur and T. E. Albrecht-Schönzart, Inorg. Chem., 2023, 62, 2513–2517 CrossRef PubMed.
- C. A. P. Goodwin, M. T. Janicke, B. L. Scott and A. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 20680–20696 CrossRef CAS PubMed.
- R. E. Jilek, N. C. Tomson, R. L. Shook, B. L. Scott and J. M. Boncella, Inorg. Chem., 2014, 53, 9818 CrossRef CAS PubMed.
- A. T. Breshears, C. L. Barnes, D. V. Wagle, G. A. Baker, M. K. Takase and J. R. Walensky, Radiochim. Acta, 2015, 103, 49–56 CrossRef CAS.
- C. Berthon, N. Boubals, I. A. Charushnikova, D. Collison, S. M. Cornet, C. Den Auwer, A. J. Gaunt, N. Kaltsoyannis, I. May, S. Petit, M. P. Redmond, S. D. Reilly and B. L. Scott, Inorg. Chem., 2010, 49, 9554–9562 CrossRef CAS PubMed.
- G. Bombieri, D. Brown and R. Graziani, J. Chem. Soc., Dalton Trans., 1975, 1873–1876 RSC.
- J. J. Kiernicki, M. Zeller and S. C. Bart, Angew. Chem., Int. Ed., 2017, 56, 1097–1100 CrossRef CAS PubMed.
- J. L. Brown, A. J. Gaunt, D. M. King, S. T. Liddle, S. D. Reilly, B. L. Scott and A. J. Wooles, Chem. Commun., 2016, 52, 5428–5431 RSC.
- J. L. Brown, E. R. Batista, J. M. Boncella, A. J. Gaunt, S. D. Reilly, B. L. Scott and N. C. Tomson, J. Am. Chem. Soc., 2015, 137, 9583–9586 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2335543–2335545 and 2345808. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01560f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |