
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bipolar polymer cathode for sodium-ion batteries†
Daniel M.
Harrison
ab,
Eric Youngsam
Kim
 ab,
Thierno B.
Rhodes
a,
Zhenzhen
Yang
c,
Mikell
Paige
ab and
Chao
Luo
*ade
ab,
Thierno B.
Rhodes
a,
Zhenzhen
Yang
c,
Mikell
Paige
ab and
Chao
Luo
*ade
aDepartment of Chemistry & Biochemistry, George Mason University, 4400 University Drive, Fairfax, VA 22030, USA
bCenter for Molecular Engineering, George Mason University, Manassas, VA 20110, USA
cChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA
dQuantum Science & Engineering Center, George Mason University, Fairfax, VA 22030, USA
eDepartment of Chemical, Environmental, and Materials Engineering, University of Miami, Coral Gables, FL 33146, USA. E-mail: cxl1763@miami.edu
First published on 17th June 2024
Abstract
A bipolar polymer cathode material, containing redox-active azo benzene and diamine moieties, was synthesized for sodium-ion batteries. The n-type azo group and p-type amine group enable a wide cutoff window with an initial capacity of 93 mA h g−1 at 50 mA g−1 and a high voltage plateau at ∼3.3 V.
The pursuit of sustainable energy storage technologies has uncovered many research paths in renewable energy. Among those, sodium-ion batteries (SIBs) have shown great promise as sustainable and renewable alternatives to lithium-ion batteries (LIBs) due to the widespread availability and low cost of sodium resources. Sharing similar monovalent chemistry, many concepts and experiences from LIBs can be extended to SIBs. However, the sodium ion's larger radius (1.0 Å vs. Li 0.7 Å), higher mass (22.99 g mol−1vs. Li 6.94 g mol−1) and higher standard redox potential (−2.71 V vs. Li −3.04 V), and lower binding energy compared to the lithium ion require new considerations in the design of SIB electrode materials.1
Organic materials are a maturing class of electrode materials that show comparable performance to conventional inorganic electrode materials that rely on rare and toxic transition metals,2 while achieving the green and affordable goals required to feasibly match global energy storage demands. These organic electrode materials (OEMs) offer high tunability through chemical modification and synthetic design to maximize performance for a wide array of applications. Several n-type functional groups have been identified as redox active centers in organic SIBs, including carbonyl, thioketone, imine, and azo groups. They show universal reversible redox activity with Li+, Na+, and K+ ions, and have been employed in both small organic molecules and high-molecular-weight polymers.3–5 Additionally, p-type anion-insertion functional groups can undergo oxidation to reversibly interact with anions.6–8 Understanding the electronic environment and microstructure of materials possessing these redox active groups is important to realizing the rational design of OEMs for SIBs.
Recently, porous organic polymers (POPs) gained considerable research interests. The high porosity affords many benefits to electrochemical performance. The capacity and rate capability are improved due to fast ion transport and charge transfer rates.9 The increased surface area enables intimate contact between active materials and electrolytes, as well as offers more active sites for redox reactions, enhancing the reaction kinetics.10–12 The high molar mass and cross-linking of POPs inhibit solubility in the electrolyte, a common challenge for fast capacity loss in organic batteries.9 So far, POPs have been employed as OEMs in SIBs.13 In addition, it was shown that azo groups can uptake two Na+ cations with redox potentials at ∼1.3 V.14–18 Alternatively, p-type functional groups, such as triphenylamine and related derivatives, can uptake anions such as the counter ion of the electrolyte salt (i.e. PF6− or ClO4−), with a higher redox potential close to 3.3 V.19–22 A number of redox-active polymers incorporating one of these groups with a variety of linking monomers have been reported. Additionally, dual-ion polymers that incorporate redox active groups in the anodic window (<1.0 V) paired with cathodic groups (>1.0 V) have also been reported.23,24 They can function as symmetric electrodes in dual-ion batteries. However, a few dual-ion polymers that function only as cathodes, with both redox plateaus above 1.0 V, have been reported in SIBs.25
In this work, a two-dimensional dual-ion porous polymer containing both triphenylamine groups and azo groups was synthesized and used as a cathode material in SIBs. A control polymer with only the amine groups was also prepared. The structures of both polymers and the proposed mechanism for the cationic and anionic redox reactions are shown in Fig. 1. The amide group was employed as a linker to connect the azo benzene/benzene moiety with the diamine moiety in TPDA-ABDC and TPDA-TCl (Fig. 1a). As shown in Fig. 1b, the azo group can reversibly react with four Na+ cations and electrons, while the N atoms in the amine groups can reversibly react with two anions and lose two electrons. These reversible reactions provide electrochemical energy for SIBs.
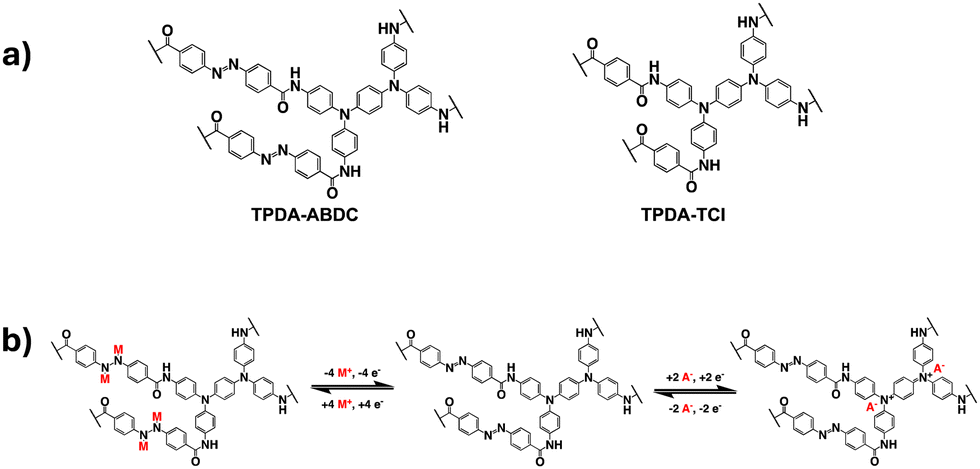 | ||
| Fig. 1 (a) The molecular structure of TPDA-ABDC and TPDA-TCl, (b) The proposed redox mechanisms for both functional groups of TPDA-ABDC, with M representing Na+ cations, and A representing PF6− anions. | ||
The synthesized polymers were characterized by Fourier-transform infrared (FTIR) and Raman spectroscopy, and scanning electron microscopy (SEM). Porosity was measured using Brunauer–Emmett–Teller (BET) method. The FTIR spectrum for TPDA-ABDC (Fig. 2a) shows absorptions for aryl rings at 1650 cm−1 and conjugated amide carbonyl groups at 1500 cm−1. The FTIR spectrum for TPDA-TCl (Fig. S2a, ESI†) is very similar because both polymers possess the same amide linker and diamine moiety with the azo linkage as the only difference. Symmetric trans azo groups do not typically absorb FTIR frequencies and thus do not appear in the spectra,26 whereas Raman spectroscopy can reveal the presence of such functional groups. The Raman spectrum for TPDA-ABDC in Fig. 2b shows a signal at 1149 cm−1, indicating the presence of the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– azo linkage, which is absent from the Raman spectrum of TPDA-TCl (Fig. S2b, ESI†). The slanted baseline is due to autofluorescence from the conjugated aromatic structures in the polymer. The pore diameter of TPDA-ABDC was determined by BET to be 18.75 nm, and the pore diameter of TPDA-TCl is 14.26 nm, showing that both polymers are mesoporous. Fig. 2c shows the N2 adsorption/desorption curve for TPDA-ABDC. The curve's shape indicates type II isotherm adsorption behavior of the polymer, showing readily reversible adsorption, and the hysteresis loop between the adsorption and desorption suggests cylindrical pore shapes.27 The morphology of the polymer was investigated by SEM (Fig. 2d) and shows micron sized rough particles. Powdered X-ray diffraction was also performed, and the results (Fig. S3, ESI†) show that neither polymer was crystalline, classifying the polymers as POPs instead of covalent organic frameworks.
N– azo linkage, which is absent from the Raman spectrum of TPDA-TCl (Fig. S2b, ESI†). The slanted baseline is due to autofluorescence from the conjugated aromatic structures in the polymer. The pore diameter of TPDA-ABDC was determined by BET to be 18.75 nm, and the pore diameter of TPDA-TCl is 14.26 nm, showing that both polymers are mesoporous. Fig. 2c shows the N2 adsorption/desorption curve for TPDA-ABDC. The curve's shape indicates type II isotherm adsorption behavior of the polymer, showing readily reversible adsorption, and the hysteresis loop between the adsorption and desorption suggests cylindrical pore shapes.27 The morphology of the polymer was investigated by SEM (Fig. 2d) and shows micron sized rough particles. Powdered X-ray diffraction was also performed, and the results (Fig. S3, ESI†) show that neither polymer was crystalline, classifying the polymers as POPs instead of covalent organic frameworks.
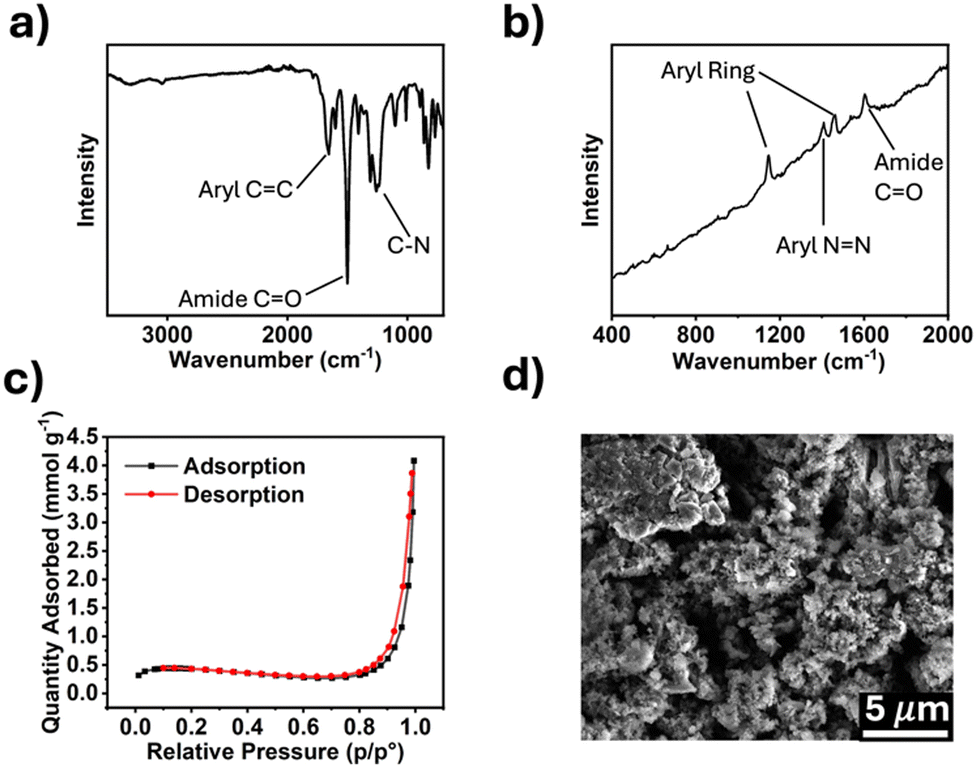 | ||
| Fig. 2 Characterization of TPDA-ABDC. (a) FTIR spectrum, (b) Raman spectrum, (c) N2 adsorption/desorption curve, (d) SEM image of TPDA-ABDC. | ||
The electrochemical performance of TPDA-ABDC was tested through galvanostatic and voltametric methods in half-cells with sodium metal as the counter electrode and a 0.3 M NaPF6 in a EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC (1:1 v/v) electrolyte system.28 This electrolyte was selected after showing a high Coulombic efficiency (>99%), which was unattainable in 1.0 M and 2.0 M NaPF6 in EC:DEC electrolytes (Fig. S4, ESI†). The galvanostatic charge and discharge curves in Fig. 3a showed a crossover point at about 3.1 V, which was attributed to the anion insertion of the triphenylamine groups. This crossover point matched with the galvanostatic charge and discharge curve graph for TPDA-TCl (Fig. S5, ESI†). Unique to the azo linked polymer was the plateau witnessed around 1.6 V on the discharge curve, indicative of the cation insertion of the azo groups. This matched well with previous literatures.18,29,30 As shown in Fig. 3b, the initial capacity was 93.7 mA h g−1, which decreased to 73.1 mA h g−1 after 100 cycles. The TPDA-TCl analogue displayed an initial capacity of 69.2 mA h g−1, which decreased to 52 mA h g−1 after 50 cycles, but the Coulombic efficiency did not reach 99% (Fig. S6, ESI†). The cycling behavior of TPDA-ABDC was further investigated with a rate performance test from 20 mA g−1 to 1 A g−1 (Fig. 3c). The capacity decreased from an initial capacity of 123.1 mA h g−1 at a current density of 20 mA g−1, to 17 mA h g−1 at 1.0 A g−1. When the current density decreased back to 20 mA g−1, it recovered to 80 mA h g−1 immediately. Cyclic voltammetry (CV) was performed on TPDA-ABDC, and the initial cycles at a 0.1 mV s−1 scan rate are shown in Fig. S7 (ESI†) (initial scans of TPDA-TCl shown in Fig. S8, ESI†). The current response of the anion insertion and extraction is witnessed above 3.0 V and is clearly reversible. Alternatively, the current response below 1.8 V, attributed to the azo redox reaction, suggests that the sodiation of the azo groups occurs at a specific potential, but desodiation occurs over a wider voltage range above 1.6 V. To explore this unique behavior, CV was run across increasing scan rates (0.1–1.0 mV s−1), and selected cycles are overlayed in Fig. 3d. Above 3 V, both the cathodic and anodic current peaks increased with the increasing polarization at higher scan rates, but below 1.8 V, only the cathodic peak current increased significantly with scan rate. The anodic region between 1.5–2.8 V shows an increase but remains primarily linear with no significant current response peaks. The natural log of the peak current values from each scan rate was plotted against the natural log of the scan rate to better determine the kinetic behavior of both redox groups in the polymer. The slope of the fitted trendline can reveal the cell's driving mechanism with a slope closer to 0.5, indicating a diffusion-controlled reaction, while a slope closer to 1.0 indicates that the surface reaction drives the kinetics. Fig. 3e shows that the cathodic and anodic slopes are 0.7753 and 0.8461, respectively, showing that both diffusion and surface reactions contribute to the reaction kinetics of the polymer cathode. Electrochemical impedance spectroscopy (EIS) was also employed to further understand the kinetics. EIS was performed on a pristine half-cell, and after cycling (Fig. S9, ESI†). The depressed semi-circle representing the interphase and charge transfer resistance decreases, suggesting improved kinetics upon cycling. Interestingly, the Warburg tail's slope steepens upon cycling, indicating additional diffusive capacity upon cycling. The kinetics of the cell was also tested by galvanostatic intermittent titration technique (GITT), and the results are shown in Fig. 3f. The overpotentials around the anion insertion redox potential of 3.2 V are 85 mV and 86 mV for discharge and charge, respectively. The overpotentials around the azo redox point at 1.8 V are notably larger, 296 mV during discharge, and 294 mV during charge, demonstrating the slow diffusion of Na+ cations within the electrode.
:PC (1:1 v/v) electrolyte system.28 This electrolyte was selected after showing a high Coulombic efficiency (>99%), which was unattainable in 1.0 M and 2.0 M NaPF6 in EC:DEC electrolytes (Fig. S4, ESI†). The galvanostatic charge and discharge curves in Fig. 3a showed a crossover point at about 3.1 V, which was attributed to the anion insertion of the triphenylamine groups. This crossover point matched with the galvanostatic charge and discharge curve graph for TPDA-TCl (Fig. S5, ESI†). Unique to the azo linked polymer was the plateau witnessed around 1.6 V on the discharge curve, indicative of the cation insertion of the azo groups. This matched well with previous literatures.18,29,30 As shown in Fig. 3b, the initial capacity was 93.7 mA h g−1, which decreased to 73.1 mA h g−1 after 100 cycles. The TPDA-TCl analogue displayed an initial capacity of 69.2 mA h g−1, which decreased to 52 mA h g−1 after 50 cycles, but the Coulombic efficiency did not reach 99% (Fig. S6, ESI†). The cycling behavior of TPDA-ABDC was further investigated with a rate performance test from 20 mA g−1 to 1 A g−1 (Fig. 3c). The capacity decreased from an initial capacity of 123.1 mA h g−1 at a current density of 20 mA g−1, to 17 mA h g−1 at 1.0 A g−1. When the current density decreased back to 20 mA g−1, it recovered to 80 mA h g−1 immediately. Cyclic voltammetry (CV) was performed on TPDA-ABDC, and the initial cycles at a 0.1 mV s−1 scan rate are shown in Fig. S7 (ESI†) (initial scans of TPDA-TCl shown in Fig. S8, ESI†). The current response of the anion insertion and extraction is witnessed above 3.0 V and is clearly reversible. Alternatively, the current response below 1.8 V, attributed to the azo redox reaction, suggests that the sodiation of the azo groups occurs at a specific potential, but desodiation occurs over a wider voltage range above 1.6 V. To explore this unique behavior, CV was run across increasing scan rates (0.1–1.0 mV s−1), and selected cycles are overlayed in Fig. 3d. Above 3 V, both the cathodic and anodic current peaks increased with the increasing polarization at higher scan rates, but below 1.8 V, only the cathodic peak current increased significantly with scan rate. The anodic region between 1.5–2.8 V shows an increase but remains primarily linear with no significant current response peaks. The natural log of the peak current values from each scan rate was plotted against the natural log of the scan rate to better determine the kinetic behavior of both redox groups in the polymer. The slope of the fitted trendline can reveal the cell's driving mechanism with a slope closer to 0.5, indicating a diffusion-controlled reaction, while a slope closer to 1.0 indicates that the surface reaction drives the kinetics. Fig. 3e shows that the cathodic and anodic slopes are 0.7753 and 0.8461, respectively, showing that both diffusion and surface reactions contribute to the reaction kinetics of the polymer cathode. Electrochemical impedance spectroscopy (EIS) was also employed to further understand the kinetics. EIS was performed on a pristine half-cell, and after cycling (Fig. S9, ESI†). The depressed semi-circle representing the interphase and charge transfer resistance decreases, suggesting improved kinetics upon cycling. Interestingly, the Warburg tail's slope steepens upon cycling, indicating additional diffusive capacity upon cycling. The kinetics of the cell was also tested by galvanostatic intermittent titration technique (GITT), and the results are shown in Fig. 3f. The overpotentials around the anion insertion redox potential of 3.2 V are 85 mV and 86 mV for discharge and charge, respectively. The overpotentials around the azo redox point at 1.8 V are notably larger, 296 mV during discharge, and 294 mV during charge, demonstrating the slow diffusion of Na+ cations within the electrode.
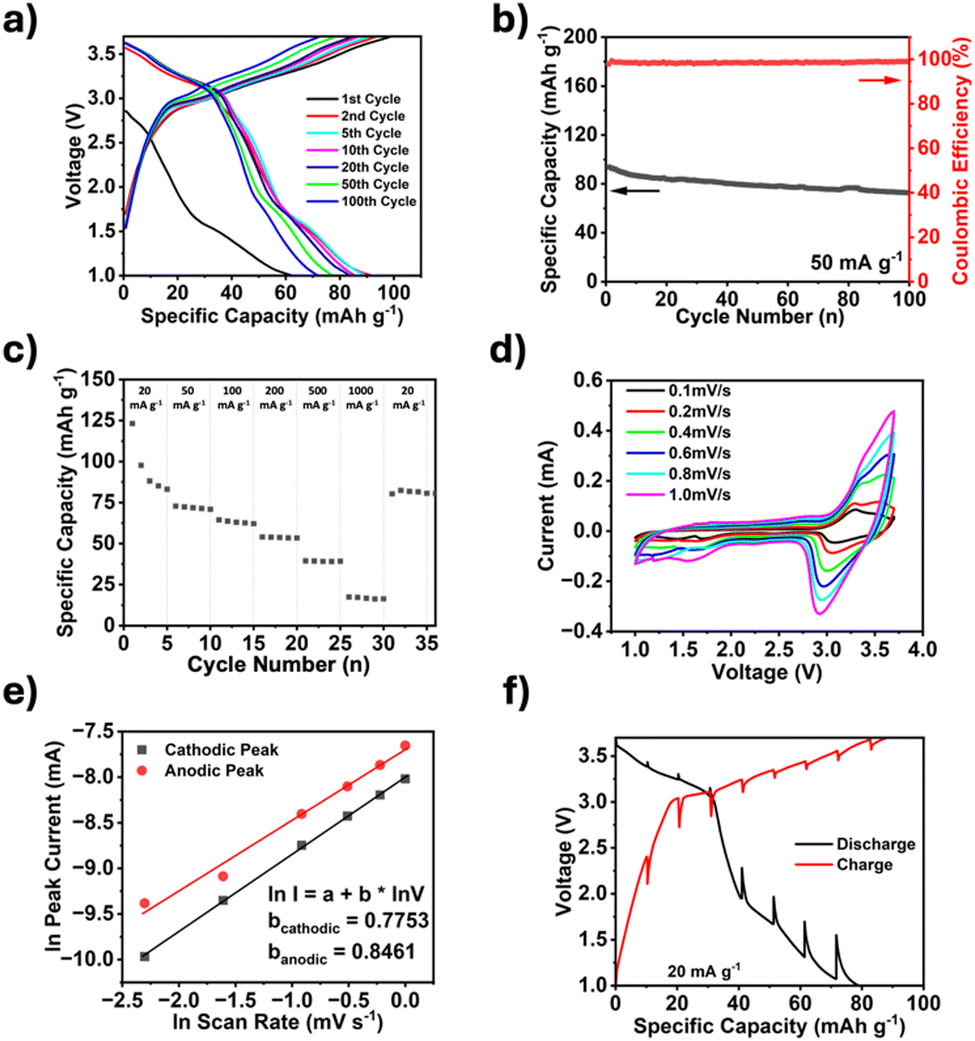 | ||
| Fig. 3 Electrochemical and kinetic tests on TPDA-ABDC. (a) Galvanostatic charge and discharge curves of select cycles at a 50 mA g−1 current density, (b) cycling performance at a current density of 50 mA g−1, (c) rate performance test from 0.02–1 A g−1, (d) cyclic voltammetry at various scan rates, (e) plot of natural log of scan rate vs. natural log of peak current, (f) GITT with a 30 min pulse time at 20 mA g−1 and 3-hour rest period. | ||
X-ray photoelectron spectroscopy (XPS) was employed to confirm reversibility of the redox reactions and interfacial chemistry (Fig. 4). The C 1s spectra shows the aryl carbon peak at 284.8 eV (C–C/CC), amide carbonyl (N–CO) peak at 288.0 eV, and aryl C–N peak at 285.7 eV. A fluorinated carbon (C–F) peak at 292.5 eV is present in the electrode spectra due to the PTFE binder. The O 1s spectra confirms the carbonyl oxygen presence with a peak at 531.5 eV. A peak at 532.7 eV appears upon cycling, attributed to C–O bonds due to the formation of cathode electrolyte interphase (CEI) by electrolyte decomposition.9 The N 1s spectra confirms the presence of the amide functional group at 401.6 eV, and triphenyl amine/aryl azo C–N peak at 400.1 eV. The azo (NN) peak at 399.7 eV disappears when the electrode is discharged to 1.0 V, and a reduced N–Na peak appears at 398.5 eV.31 The azo peak reappears upon charging the electrode to 2.2 V, with a diminished sodiated azo peak remaining that matches the slower desodiation rate in the CV in Fig. 3d. This confirms the reversible redox reaction of the azo group. The PF6− insertion is also confirmed by the N 1s spectra by the appearance of the oxidized nitrogen (N+⋯PF6−) peak at 400.9 eV in the fully charged electrode,9,32 that subsequently disappears after discharging. This confirms the reversibility of both cationic and anionic redox reactions of TPDA-ABDC.
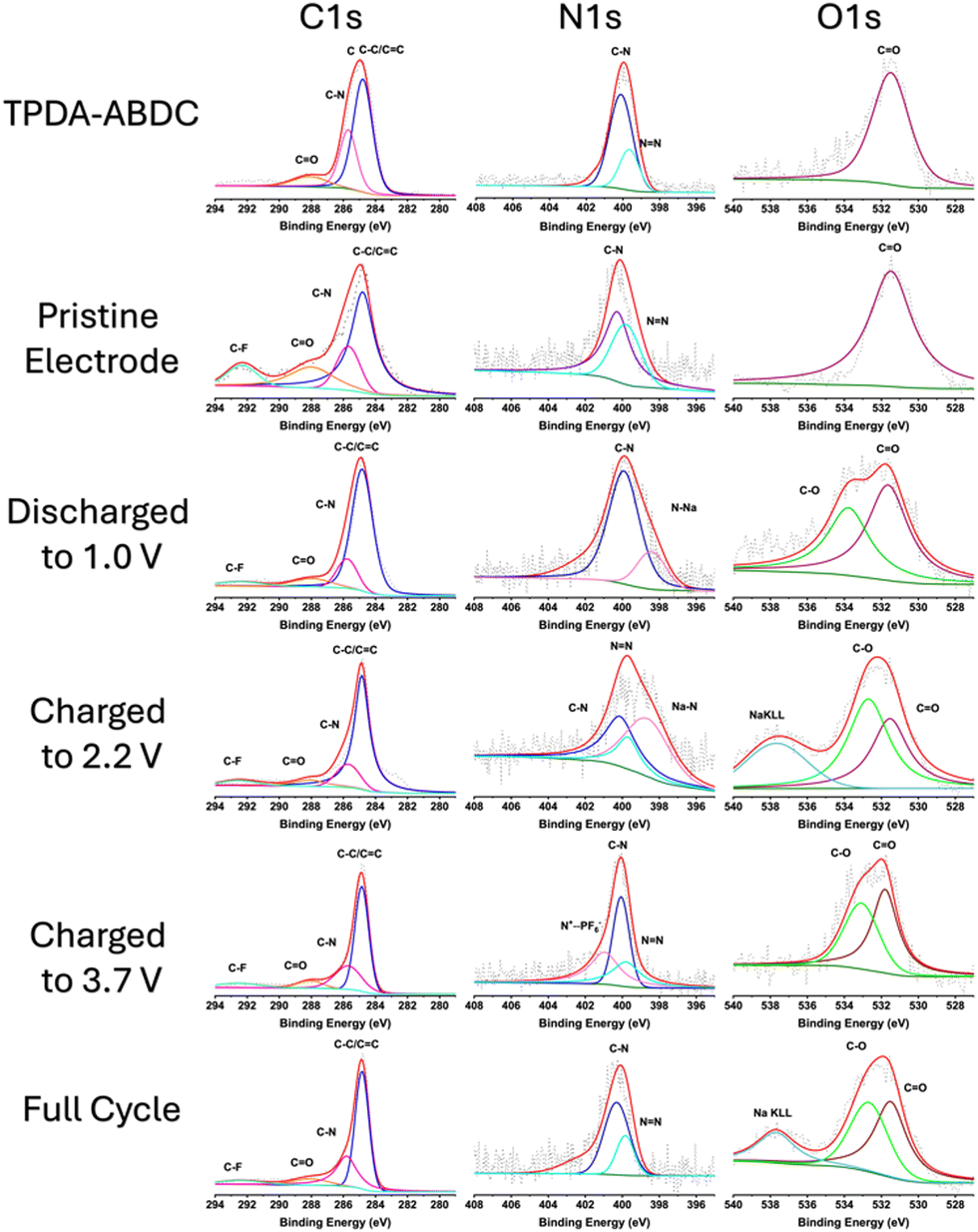 | ||
| Fig. 4 XPS characterization of pure TPDA-ABDC, pristine and cycled TPDA-ABDC electrodes at various states of discharge and charge. | ||
Post-cycling characterization of TPDA-ABDC was also performed to study its structure stability after cycling. FTIR spectroscopy was employed to confirm the chemical stability of pristine and cycled electrodes. Fig. 5a shows consistent peaks through 50 cycles, highlighting the stability of TPDA-ABDC upon cycling. The surface morphology of the electrodes was investigated by SEM. The pristine electrode morphology (Fig. 5b) stays consistent in the cycled electrodes after 1 and 5 cycles (Fig. 5c and d), showing that no significant changes occur in the bulk electrode composite throughout cycling. These results confirm the structure stability of the polymer cathode upon cycling, demonstrating that TPDA-ABDC is a promising polymer cathode for SIBs.
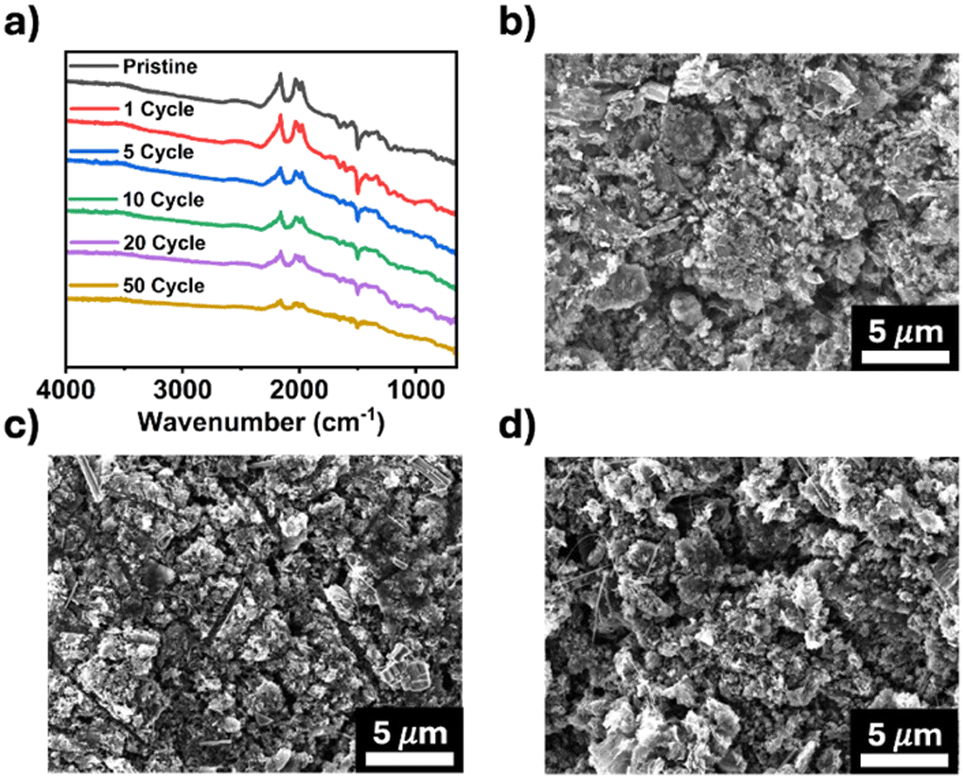 | ||
| Fig. 5 Post-cycling characterization of TPDA-ABDC, (a) FTIR spectra of TPDA-ABDC before and after cycling, (b) SEM image of a pristine electrode, (c) SEM image after 1 cycle, (d) SEM image after 5 cycles. | ||
In summary, a bipolar organic polymer (TPDA-ABDC), containing redox-active azo and triphenylamine groups, was designed and synthesized. An analogue polymer, TPDA-TCl, lacking the azo functional group, was also studied as a control sample. The azo group was shown to undergo reversible redox with sodium ions, as well as increasing the pore size of the TPDA-ABDC compared to its analogue, which enables higher capacity, cycling stability, and Coulombic efficiency. The overall results show that the combination of the n-type azo group and p-type amine group in the porous polymer enables a polymer cathode with an initial capacity of 93 mA h g−1 at 50 mA g−1 and a high-voltage plateau at ∼3.3 V in SIBs. Moreover, the polymer cathode exhibits a stable cycle life of 100 cycles and high rate capability up to 1.0 A g−1, demonstrating a promising cathode for SIBs. This work provides a structural design strategy by combining the n-type and p-type functional groups in the porous polymer to achieve high-performance cathodes for affordable and sustainable SIBs.
This work was supported by the US National Science Foundation Award No. 2419947. The authors also acknowledge the support from the Quantum Science & Engineering Center and College of Science at George Mason University, as well as Dr. Tao Gao and Yunan Qin at the University of Utah. We gratefully acknowledge support from the Post Test Facility at Argonne National Laboratory, which is operated for the DOE Vehicle Technologies Office (VTO) by UChicago Argonne, LLC, under contract number DE-AC02-06CH11357.
Data availability
Data for this article, including raw data and figures, are available upon reasonable request to the corresponding author. The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Gong, C. Wei, Z. Liang and Y. Tang, Small Sci., 2021, 1, 2100014 CrossRef CAS.
- P. Gupta, S. Pushpakanth, M. A. Haider and S. Basu, ACS Omega, 2022, 7, 5605 CrossRef CAS PubMed.
- J. J. Shea and C. Luo, ACS Appl. Mater. Interfaces, 2020, 12, 5361 CrossRef CAS PubMed.
- M. G. Mohamed, S. U. Sharma, C.-H. Yang, M. M. Samy, A. A. K. Mohammad, S. V. Chaganti, J.-T. Lee and S. Wei-Kuo, ACS Appl. Energy Mater., 2021, 4, 14628 CrossRef CAS.
- M. Mohammadiroudbari, K. Qin and C. Luo, Batteries Supercaps, 2022, 5, e202200021 CrossRef CAS.
- A. E. Lakraychi, F. Dolhem, A. Vlad and M. Becuwe, Adv. Energy Mater., 2021, 11, 2101562 CrossRef CAS.
- H. Wang, Q. Li, Q. Wu, Z. Si, L. Xiaoling, X. Liang, H. Wang, L. Sun, W. Shi and S. Song, Adv. Energy Mater., 2021, 11, 2100381 CrossRef CAS.
- L. Xu, S. Zhang, P. Guo and C. Su, ChemistrySelect, 2021, 6, 4725 CrossRef CAS.
- T. Li, W. Zhu, R. Shen, H.-Y. Wang, W. Chen, S.-J. Hao, Y. Li, Z.-G. Gu and Z. Li, New J. Chem., 2018, 42, 6247 RSC.
- N. Xu, S. Mei, Z. Chen, Y. Dong, W. Li and C. Zhang, Biochem. Eng. J., 2020, 395, 124975 CAS.
- K. S. Weeraratne, A. A. Alzharani and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2019, 11, 23520 CrossRef CAS PubMed.
- X. Liu, C. Liu, W. Lai and W. Huang, Adv. Mater. Technol., 2020, 5, 2000154 CrossRef CAS.
- F. Y. Chou, J. C. Tang, H. Y. Lee, J. C. Lee, S. Ratchahat, T. H. Chen and W. Kaveevivitchai, ACS Appl. Energy Mater., 2020, 3, 11300 CrossRef CAS.
- C. Luo, G.-L. Xu, X. Ji, S. Hou, L. Chen, F. Wang, J. Jiang, Z. Chen, Y. Ren, K. Amine and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 2879 CrossRef CAS PubMed.
- C. Luo, X. Ji, S. Hou, N. Edison, X. Fan, Y. Liang, T. Deng, J. Jiang and C. Wang, Adv. Mater., 2018, 30, 1706498 CrossRef PubMed.
- C. Wu, M. Hu, X. Yan, G. Shan, J. Liu and J. Yang, Energy Storage Mater., 2021, 36, 347 CrossRef.
- T. Shimizu, N. Tanifuji and H. Yoshikawa, Angew. Chem., Int. Ed., 2022, 61, e202206093 CrossRef CAS PubMed.
- C. Luo, O. Borodin, X. Ji, S. Hou, K. J. Gaskell, X. Fan, J. Chen, T. Deng, R. Wang, J. Jiang and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2004 CrossRef CAS PubMed.
- L. Mo, G. Zhou, P. Ge, Y.-E. Miao and T. Liu, Sci. China Mater., 2021, 65, 32 CrossRef.
- W. Ni, J. Cheng, X. Li, G. Gu, L. Huang, Q. Guan, D. Yuan and B. Wang, RSC Adv., 2015, 5, 9221 RSC.
- K. Yamamoto, D. Suemasa, K. Masuda, K. Aita and T. Endo, ACS Appl. Mater. Interfaces, 2018, 10, 6346 CrossRef CAS PubMed.
- W. Deng, X. Liang, X. Wu, J. Qian, Y. Cao, X. Ali, J. Feng and H. Yang, Sci. Rep., 2013, 3, 2671 CrossRef PubMed.
- G. Dai, Y. He, Z. Niu, P. He, C. Zhang, Y. Zhao and H. Zhou, Angew. Chem., Int. Ed., 2019, 58, 9902 CrossRef CAS PubMed.
- Z. Sun, K. Zhu, P. Liu, H. Li and L. Jiao, Adv. Funct. Mater., 2021, 31, 2107830 CrossRef CAS.
- S. Chen, T. Jia, G. Zhou, C. Zhang, Q. Hou, Y. Wang, S. Luo, G. Shi and Y. Zeng, J. Electrochem. Soc., 2019, 166, A2543 CrossRef CAS.
- A. V. Zemskov, G. N. Rodionova, Y. G. Tuchin and V. V. Karpov, J. Appl. Spectrosc., 1988, 49, 1020 CrossRef.
- R. Bardestani, G. S. Patience and S. Kaliaguine, Can. J. Chem. Eng., 2019, 97, 2781 CrossRef CAS.
- Y. Li, Y. Yang, Y. Lu, Q. Zhou, X. Qi, Q. Meng, X. Rong, L. Chen and Y.-S. Yu, ACS Energy Lett., 2020, 5, 1156 CrossRef CAS.
- T. Shimizu, T. Mameuda, H. Toshima, R. Akiyoshi, Y. Kamakura, K. Wakamatsu, D. Tanaka and H. Yoshikawa, ACS Appl. Energy Mater., 2022, 5, 5191 CrossRef CAS.
- J. Wang, Y. Tong, W. Huang and Q. Zhang, Batteries Supercaps, 2022, 6, e202200413 CrossRef.
- E. Y. Kim, M. Mohammadiroudbari, F. Chen, Z. Yang and C. Luo, ACS Nano, 2024, 18, 4159–4169 CrossRef CAS PubMed.
- Z. Peng, X. Yi, Z. Liu, J. Shang and D. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 14578–14585 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01479k |
| This journal is © The Royal Society of Chemistry 2024 |