
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCell viability imaging in tumor spheroids via DNA binding of a ruthenium(II) light-switch complex†
Vadde
Ramu
 *a,
Lukas S.
Wijaya
b,
Nataliia
Beztsinna
a,
Corjan
Van de Griend
a,
Bob
van de Water
b,
Sylvestre
Bonnet
*a and
Sylvia E.
Le Dévédec
*b
*a,
Lukas S.
Wijaya
b,
Nataliia
Beztsinna
a,
Corjan
Van de Griend
a,
Bob
van de Water
b,
Sylvestre
Bonnet
*a and
Sylvia E.
Le Dévédec
*b
aLeiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: ramuvadde1@gmail.com; bonnet@chem.leidenuniv.nl
bLeiden Academic Center for Drug Research, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: s.e.ledevedec@lacdr.leidenuniv.nl
First published on 13th May 2024
Abstract
The famous ‘‘light-switch’’ ruthenium complex [Ru(bpy)2(dppz)](PF6)2 (1) has been long known for its DNA binding properties in vitro. However, the biological utility of this compound has been hampered by its poor cellular uptake in living cells. Here we report a bioimaging application of 1 as cell viability probe in both 2D cells monolayer and 3D multi-cellular tumor spheroids of various human cancer cell lines (U87, HepG2, A549). When compared to propidium iodide, a routinely used cell viability probe, 1 was found to enhance the staining of dead cells in particular in tumor spheroids. 1 has high photostability, longer Stokes shift, and displays lower cytotoxicity compared to propidium iodide, which is a known carcinogenic. Finally, 1 was also found to displace the classical DNA binding dye Hoechst in dead cells, which makes it a promising dye for time-dependent imaging of dead cells in cell cultures, including multi-cellular tumor spheroids.
Drug discovery is an expensive and lengthy process that proceeds via several stages from target identification to lead discovery and optimization, preclinical demonstration, and clinical trials that finally may lead to approval for use in humans.1–3 A noteworthy step in this procedure is high-throughput screening (HTS) of small compounds for lead identification.4 In this process dyes such as propidium iodide (PI), SYTOX green DNA-Responsive Azo-Based Quantum 7 (DRAQ7), or 7AAD (7-amino-actinomycin D) are commonly used for cell viability assay. The main flaws of such fluorophores are their limited stability in aqueous solutions, which involves frequent preparation of stock solutions, and their sub-optimal photostability in cells, which prevents long-time-scale imaging. Also, their small Stokes’ shifts demand a precise selection of excitation wavelengths and the use of cut-off filters to eliminate scattering by the excitation beam and the auto-fluorescence from the samples themselves. Besides, small Stokes’ shifts limit multicolour labelling in confocal microscopy and flow cytometry. Finally, the short nanosecond timescale of these probes hinders the development of cellular viability assays relying on time-resolved emission measurements using “flash” excitation methods in standard microplate fluorometers. In addition, most cell-based HTS is being carried out in two-dimensional (2D) monolayer cell cultures5 or more complex three-dimensional (3D) co-cultures.6 It has been suggested that the oversimplistic character of 2D monolayer cell viability assays may partially explain the high failure percentage in drug development.7,8 Compared with 2D monolayer cell cultures, 3D multi-cellular tumor spheroids (MCTS) more accurately simulate many features of real human solid tumors in vivo, such as their spatial architecture, physiological responses, secretion of soluble mediators, gene expression patterns, and drug resistance mechanisms.9 The use of MCTS should allow superior predictability of the efficacy and toxicity of new drug molecules that, in turn, may reduce the erosion rate of new molecular medicines under progress.10 To establish routine MCTS assays, improved protocols are essential for imaging cell death within a developing 3D spheroid. Such a requirement necessitates the development of more photostable molecular dyes that can differentiate in real-time living from deceased cells when observing a 3D tumour spheroid under a microscope.
Ruthenium polypyridyl complexes have been widely used in bioimaging due to their excitability in the visible region, low photobleaching rate, high Stokes shifts and lifetimes, relatively low toxicity, and excellent chemical (photo)stability.11–17 The triplet character of their excited states, however, is a potential issue for bioimaging, as it may lead to singlet oxygen generation and phototoxicity, thereby hampering long-term imaging of the living process.18,19 TLD-1433, for example, has entered phase II clinical trials due to its photodynamic therapeutic properties and could not be used for bioimaging.20 On the other hand, [Ru(bpy)2(dppz)](PF6)2 (1, Scheme 1a; bpy = 2,2′-bipyridine, dppz = dipyrido[3,2-a:2′,3′-c]phenazine) is well-known for being non-luminescent in aqueous solution but displaying bright red luminescence following intercalation into the double-helix of DNA, which is characterized by an >104 enhancement of its phosphorescence quantum yield.21 This well-known effect is called the “light switch” effect in the literature and it has been thoroughly investigated in the last two decades.22–25 These studies have shown that 1 has poor cellular internalization, and that the compound needs to be chemically modified for cell viability staining in 2D cell cultures.26,27 Lincoln and others proposed analogous RuII(dppz) complexes for nuclear DNA staining in dead or fixed cells that required photo-activation or ion pairing to be taken up in cells.25,28 In the literature, alternative methods were proposed to enhance the cell penetration of the ruthenium light switch complex by peptide vectorization leading them to nucleus localization.32 Rajendiran, Jimenez, Gill, and others have also reported that 1 and some derivatives (2,2′-bipyridine, 1,10-phenanthroline) could be used as staining agents for nonviable cells in 2D monolayers which allowed to dramatically improve flowcytometry analysis for example.29–31
 | ||
| Scheme 1 Chemical structure of the light-switch [Ru(bpy)2(dppz)](PF6)2 complex 1 (a) and propidium iodide (PI) (b). | ||
However, the use of ruthenium complexes such as 1 for the imaging of MCTS has been poorly investigated. In particular, it is not clear whether the metal complexes can penetrate the slowly proliferating core of a spheroids, or whether it would remain in their highly proliferative periphery. In this article, we addressed this question by comparing the penetration depth of 1 in U87 (glioblastoma) brain cancer spheroids with that of the classical organic dye PI. We also report the application of complex 1 as cell viability probe in living (hence, non-fixed) 3D U87 tumor spheroids. 1 is known to localize in the cytoplasm and not to be able to penetrate inside the nucleus of living cells.25 However, we found that in cells with compromised membrane, 1 could enter the nucleus and stain nucleic acids. During programmed cell death, 1 was found to displace the classical DNA-staining Hoechst dye, However, propidium iodide was not displaced by Hoechst under similar conditions.
1 was prepared as reported.21 First, to identify an ideal concentration for bioimaging experiments where 1 is not cytotoxic we measured the effective cell growth inhibition concentration (EC50) of 1 in HepG2 (liver cancer) and U87 (glioblastoma) MCTS with size ≥300 μM using the Celltiter-Glo™ 3D cell viability assay. This assay is a measure of the ATP concentration and hence a good indicator of the degree of metabolic activity in cells. Cisplatin was used as cytotoxic control. The EC50 values for 1 in HepG2 and U87 cells were determined to be ≥70 μM, while for cisplatin the EC50 values were found to be ≥2 (Table S1, ESI†). We observed no synergistic cytotoxicity effect when both 1 (50 μM) and cisplatin (50 μM) were added to U87 cells (Fig. S1, ESI†). Based on these findings, treating cells with 1 at a concentration of 50 μM resulted in 100% cell viability. Consequently, we employed this lower concentration (50 μM) in all subsequent imaging studies.
To investigate the cellular internalization and dead cell staining properties of 1 in 2D cell monolayers, The commercially available dyes propidium iodide (PI), and Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 343 were used unless otherwise mentioned in studies. We performed a live cell time-lapse imaging experiment overnight (0–16 hours) in three different cell lines, i.e. HepG2, U87, and A549 (epithelial carcinoma) cells, and compared it with propidium iodide. Cell death was induced with cisplatin and followed by staining with either 1 or PI, combined with the DNA-staining dye Hoechst 33343 to detect single cells (Fig. S2, ESI†).33Fig. 1a and b shows the magnified view of A549 cells nuclei. Interestingly, while the Hoechst signal remained strong and unchanged in the nuclei of the dying cells co-stained with PI, it decayed over time in the nuclei of the dying cells co-stained with 1 (Fig. 1a and c). As a result, both PI and Hoechst signals colocalized during the cell death process (overlay blue and red is pink) while 1 replaced the Hoechst signal (only red signal), also see time-lapse Video S1(PI and Hoechst) and S2 (ESI†) (complex 1 and Hoechst).
343 were used unless otherwise mentioned in studies. We performed a live cell time-lapse imaging experiment overnight (0–16 hours) in three different cell lines, i.e. HepG2, U87, and A549 (epithelial carcinoma) cells, and compared it with propidium iodide. Cell death was induced with cisplatin and followed by staining with either 1 or PI, combined with the DNA-staining dye Hoechst 33343 to detect single cells (Fig. S2, ESI†).33Fig. 1a and b shows the magnified view of A549 cells nuclei. Interestingly, while the Hoechst signal remained strong and unchanged in the nuclei of the dying cells co-stained with PI, it decayed over time in the nuclei of the dying cells co-stained with 1 (Fig. 1a and c). As a result, both PI and Hoechst signals colocalized during the cell death process (overlay blue and red is pink) while 1 replaced the Hoechst signal (only red signal), also see time-lapse Video S1(PI and Hoechst) and S2 (ESI†) (complex 1 and Hoechst).
 | ||
| Fig. 1 Time-dependent displacement of Hoechst by 1 or propidium iodide (PI) in dying A549 cells treated with cisplatin (50 μM) for 16 h. A549 cells stained with either (a) Hoechst and 1 or (c) Hoechst and PI (20 μM). Scale bar is 10 μm. Quantification of the total fluorescence intensities in the doubled labeled nuclei with Hoechst and 1(c), or Hoechst and PI (d). Error bars indicate SEM for about 10 nuclei. | ||
On the other hand, in cells co-stained with 1, the average signal intensity of Hoechst decreased while the signal of 1 increased overall, in 2D monolayers 1 entered the cells during late cell death when the cell membrane became compromised, and displaced Hoechst (Fig. S1 and Video S2, ESI†). This unprecedented observation makes 1 a good dead cell staining agent. To explain this observation, we recorded the emission spectra of Hoechst (λex 350 nm) with pUC19 DNA in the presence of either 1 or PI in phosphate saline buffer at pH = 7.4 (Fig. S4, ESI†) in cell-free conditions. The emission intensity of the Hoechst–DNA complex at λem 480 nm was only quenched by increasing concentrations of 1 (0–80 μM) without the appearance of any new band (Fig. S4A, ESI†), while in the presence of increasing PI concentration a new emission band was detected at λem 610 nm (Fig. S4b, ESI†). We hypothesize that the latter observation is a consequence of Förster resonance energy transfer (FRET) between Hoechst and PI. For 1, there seems to be no FRET, which corroborates the displacement of the Hoechst dye by 1 as observed by microscopy imaging of the dying A549 cells.
So far, ruthenium complexes including the dppz ligand were shown to be able to displace ethidium bromide interacting with CT-DNA, but these studies did not consider 3D tumour spheroids.34 To see how 1 behaved as an imaging agent in a 3D context we investigated the time evolution of Hoechst displacement in U87 tumor spheroids pre-treated with cisplatin (50 μM) for 40 h (Fig. 2). The minimum amount of DMSO (<0.5%) was used to dissolve 1. A vehicle control experiment was conducted to prove that DMSO alone did not induce any cell death in 3D tumor spheroids. The CellProfiler software was used to quantitatively measure the different fluorescent signals from the confocal images. The left side graphs in Fig. 2c and d represent the normalized number of nuclei detected in the Hoechst channel vs. time, using the t = 0 nuclei number as a reference. The right-side graphs in Fig. 2c and d represent the time evolution of the intensity ratio of red vs. blue emission, which represents a quantification of cell death vs. time. Under the control conditions, there was a slight increase in the number of cells within the spheroids, a trend that also appeared evident in the spheroids treated with cisplatin, as observed through PI staining (Fig. S5, ESI†). In reality, image analysis detected all the stained nuclei including healthy and dying ones since we use the masking of the Hoechst signal. When staining with 1 we did not detect any dying cells through masking of the Hoechst dye, which is visible in the decreasing curve of the cisplatin-exposed spheroids-(Fig. 2c, yellow line). The initial increase in the red/blue emission ratio (Fig. 2c right) was attributed to the background emission. The time-lapse images of the selected regions of U87 spheroids in Fig. S6 (ESI†) agree with the observation made in 2D monolayers: 1 displaces the Hoechst dye in compromised/dead cells. Although 1 and PI interact with DNA via intercalation,6 the displacement of Hoechst was not observed in the case of PI, leading to double staining.
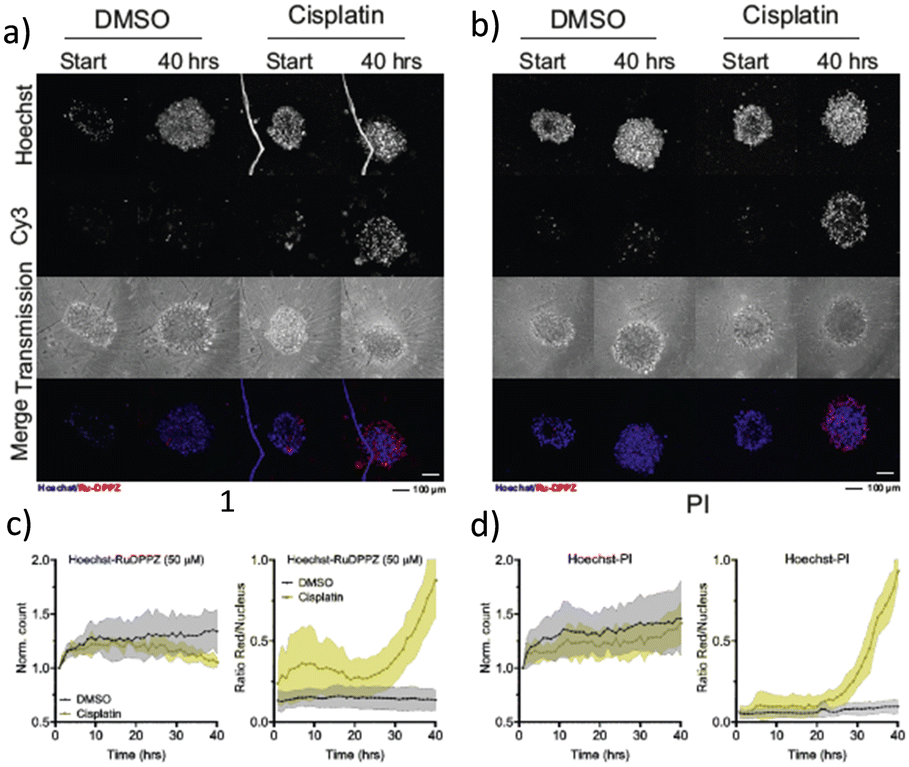 | ||
| Fig. 2 Live imaging of U87 tumor spheroids exposed to cisplatin using Hoechst or 1 or PI dyes. Cancer spheroids were treated with either DMSO or Cisplatin (50 μM) and stained with Hoechst and 1 (50 μM) (a) or Hoechst and propidium iodide (20 μM) (b). Scale: 100 μm. Quantification includes (i) normalized nuclear count vs. time-based on Hoechst staining and (ii) Cy3 signal ratio over the nuclear signal for complex 1 staining (c) or PI staining (d). | ||
Furthermore, the z-stack image (Fig. 3a) of spheroids stained with 1 and Hoechst revealed a distinct gradient of multiple layers in U87 spheroids with the partial displacement of Hoechst by 1 at the centre and complete displacement at the proliferative cells with dominated red emission. However, the z-stack image (Fig. 3b) of PI staining of U87 spheroid was shown the complementary pink colour due to the superimposition of both dyes in the dead cells pretreated with cisplatin. This unique characteristic behaviour of 1 makes it a potential candidate for general cell viability assessment.
 | ||
| Fig. 3 Z-stack image of cisplatin treated U87 tumor spheroids stained with (a) 1 (50 μM) and (b) PI (20 μM) for 40 h. (a) The yellow circle shows the separation of senescent cells from proliferative cells where Hoechst was completely displaced by 1 in dead cells. (b) Double staining of cells in U87 tumor spheroid by Hoechst and PI. Scale bar: 100 μm. | ||
Next, we examined the penetration depth of 1 and PI in the cultured 3D spheroids of HepG2 and U87 cells (Fig. 4). Due to the limitation of confocal microscopy in visualising the deeper layers of the spheroids,35 we fixed them with paraformaldehyde and prepared cryomolds using a literature procedure.36,37 We then performed cryo-sectioning of the spheroids with the Leica CM3050S cryostat.38,39 The fluorescence signal of 1 was still detectable by imaging the spheroids coupes with a confocal microscope (Fig. 4c). Interestingly, HepG2 spheroids displayed a hypoxic core which was detected by both PI and 1 but while PI seems to label only necrotic cells (Fig. 4e), 1 seem to label both apoptotic and necrotic cells (Fig. 4a). Furthermore, upon fixation and sectioning, it was interesting to observe that only the fluorescent signal of 1 was detectable and not PI anymore (Fig. 4c and g).
 | ||
| Fig. 4 Confocal images of live HepG2 and U87 spheroids stained with 1 ((a)–(d), untreated with cisplatin) and PI (e)–(h). The sliced spheroid coupes showed in bottom images (c), (d), (g), and (h). | ||
In this work, we explored the application of the light-switch complex 1 as a cell viability-detecting reagent in 2D and 3D cell cultures. In 2D monolayers of HepG2, U87, and A549 cells, 1 successfully entered the nucleus of dying cells and displaced the classical DNA-binding dye Hoechst. This work represents the first experimental observation of a ruthenium complex displacing Hoechst in dying cancer cells. The fluorescence titration results suggested that the FRET phenomenon was observed when pUC19 DNA was treated with Hoechst and PI, but not when 1 was used instead, which was attributed to the displacement of Hoechst by 1. This displacement also worked in 3D multicellular U87 and HepG2 tumor spheroids, in which the ruthenium complex could be used to quantify the time evolution of cell death upon cisplatin treatment. Overall, our results demonstrate that the well-known advantages of 1 for in vitro imaging, i.e., its high photostability, low cytotoxicity, low cell membrane penetration properties in living cells, and excellent DNA-interaction properties in dead cells, allows safe and automated time-resolved cell viability quantification in 3D tumour spheroids.
The authors gratefully acknowledge the Leiden University Cell Observatory for their support & assistance in this work. B. vd W. acknowledges the European Union Horizon's 2020 research and innovation program for funding L. W. through RISK-HUNT3R (grant agreement No 964537). S. B. acknowledges ERC for a Proof-of-Concept grant (HypoRuLight, GA 768166).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. L. Dahlin, J. Inglese and M. A. Walters, Nat. Rev. Drug. Discov., 2015, 14, 279 CrossRef CAS PubMed.
- J. Jabs, F. M. Zickgraf, J. Park, S. Wagner, X. Jiang, K. Jechow, K. Kleinheinz, U. H. Toprak, M. A. Schneider, M. Meister, S. Spaich, M. Sutterlin, M. Schlesner, A. Trumpp, M. Sprick, R. Eils and C. Conrad, Mol. Syst. Biol., 2017, 13, 955 CrossRef PubMed.
- R. C. Mohs and N. H. Greig, Alzheimers. Dement., 2017, 3, 651 CrossRef PubMed.
- S. K. Sukumaran, R. Ranganatha and S. Chakravarthy, Int. J. High Thr. Scre., 2016, 6, 1 Search PubMed.
- J. van Arensbergen, L. Pagie, V. D. FitzPatrick, M. de Haas, M. P. Baltissen, F. Comoglio, R. H. van der Weide, H. Teunissen, U. Vosa, L. Franke, E. de Wit, M. Vermeulen, H. J. Bussemaker and B. van Steensel, Nat. Genet., 2019, 51, 1160 CrossRef CAS PubMed.
- D. Lv, Z. Hu, L. Lu, H. Lu and X. Xu, Oncol. Lett., 2017, 14, 6999 Search PubMed.
- O. Sirenko, T. Mitlo, J. Hesley, S. Luke, W. Owens and E. F. Cromwell, Assay Drug Dev. Technol., 2015, 13, 402 CrossRef CAS PubMed.
- R. Demuynck, I. Efimova, A. Lin, H. Declercq and D. V. Krysko, Cells, 2020, 9, 1 CrossRef PubMed.
- F. Mittler, P. Obeid, A. V. Rulina, V. Haguet, X. Gidrol and M. Y. Balakirev, Front. Oncol., 2017, 7, 293 CrossRef PubMed.
- J. Hagemann, C. Jacobi, M. Hahn, V. Schmid, C. Welz, S. Schwenk-Zieger, R. Stauber, P. Baumeister and S. Becker, Anticancer Res., 2017, 37, 2201 CrossRef CAS PubMed.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179 RSC.
- R. C. Curley, C. S. Burke, K. S. Gkika, S. Noorani, N. Walsh and T. E. Keyes, Inorg. Chem., 2023, 62, 13089 CrossRef CAS PubMed.
- S. Singh, M. Varma, B. Shravage, P. Kulkarni and A. Kumbhar, J.Chem. Sci., 2021, 133, 89 CrossRef CAS.
- A. Gandioso, E. Izquierdo-Garcia, P. Mesdom, P. Arnoux, N. Demeubayeva, P. Burckel, B. Saubamea, M. Bosch, C. Frochot, V. Marchan and G. Gasser, Chem. – Euro. J., 2023, 29, e202301742 CrossRef CAS PubMed.
- V. Ramu, F. Ali, N. Taye, B. Garai, A. Alam, S. Chattopadhyay and A. Das, J. Mater. Chem. B, 2015, 3, 7177 RSC.
- L. Blackmore, R. Moriarty, C. Dolan, K. Adamson, R. J. Förster, M. Devocelle and T. E. Keyes, Chem. Commun., 2013, 49, 2658 RSC.
- C. E. Elgar, N. A. Yusoh, P. R. Tiley, N. Kolozsvari, L. G. Bennett, A. Gamble, E. V. Pean, M. L. Davies, C. J. Staples, H. Ahmad and M. R. Gill, J. Am. Chem. Soc., 2023, 145, 1236 CrossRef CAS PubMed.
- Y. Sun, S. N. Collins, L. E. Joyce and C. Turro, Inorg. Chem., 2010, 49, 4257 CrossRef CAS PubMed.
- E. Wachter, D. Moya, S. Parkin and E. C. Glazer, Chem. – Eur. J., 2016, 22, 550 CrossRef CAS PubMed.
- S. Chamberlain, H. D. Cole, J. Roque, 3rd, D. Bellnier, S. A. McFarland and G. Shafirstein, Pharmaceuticals, 2020, 13, 137 CrossRef CAS PubMed.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 2002, 112, 4960 CrossRef.
- M. G. Walker, V. Ramu, A. Meijer, A. Das and J. A. Thomas, Dalton Trans., 2017, 46, 6079 RSC.
- Y. Liu, A. Chouai, N. N. Degtyareva, D. A. Lutterman, K. R. Dunbar and C. Turro, J.Am. Chem. Soc., 2005, 127, 10796 CrossRef CAS PubMed.
- E. Wachter, B. S. Howerton, E. C. Hall, S. Parkin and E. C. Glazer, Chem. Commun., 2014, 50, 311 RSC.
- B. Z. Zhu, X. J. Chao, C. H. Huang and Y. Li, Chem. Sci., 2016, 7, 4016 RSC.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46 CrossRef CAS PubMed.
- F. R. Svensson, J. Andersson, H. L. Amand and P. Lincoln, J. Biol. Inorg. Chem., 2012, 17, 565 CrossRef CAS PubMed.
- F. R. Svensson, M. Matson, M. Li and P. Lincoln, Biophys. Chem., 2010, 149, 102 CrossRef CAS PubMed.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- V. Rajendiran, M. Palaniandavar, V. S. Periasamy and M. A. Akbarsha, J. Inorg. Biochem., 2010, 104, 217–220 CrossRef CAS PubMed.
- M. E. Jiménez-Hernández, G. Orellana, F. Montero and M. T. Portolés, Photochem. Photobiol., 2000, 72, 28–34 CrossRef.
- C. S. Burke, A. Byrne and T. E. Keyes, Angew. Chem., Int. Ed., 2018, 57, 12420–12424 CrossRef CAS PubMed.
- M. Rosenberg, N. F. Azevedo and A. Ivask, Sci. Rep., 2019, 9, 6483 CrossRef PubMed.
- F. D. Abreu, T. d F. Paulo, M. H. Gehlen, R. A. Ando, L. G. F. Lopes, A. C. S. Gondim, M. A. Vasconcelos, E. H. Teixeira, E. H. S. Sousa and I. M. M. de Carvalho, Inorg. Chem., 2017, 56, 9084–9096 CrossRef CAS PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839 CrossRef CAS PubMed.
- J. M. Santos, S. P. Camoes, E. Filipe, M. Cipriano, R. N. Barcia, M. Filipe, M. Teixeira, S. Simoes, M. Gaspar, D. Mosqueira, D. S. Nascimento, O. P. Pinto-do, P. Cruz, H. Cruz, M. Castro and J. P. Miranda, Stem Cell Res. Ther., 2015, 6, 90 CrossRef PubMed.
- M. Upreti, A. Jamshidi-Parsian, N. A. Koonce, J. S. Webber, S. K. Sharma, A. A. Asea, M. J. Mader and R. J. Griffin, Transl. Oncol., 2011, 4, 365 CrossRef PubMed.
- S. Walenta, J. Doetsch, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Histochem. Cytochem., 2000, 48, 509 CrossRef CAS PubMed.
- E. Leary, C. Rhee, B. T. Wilks and J. R. Morgan, SLAS Technol., 2018, 23, 231 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01425a |
| This journal is © The Royal Society of Chemistry 2024 |