
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective binding of c-MYC G-quadruplex caged in a dsDNA by a hemopeptide†
Leen
Massalha
 ,
Adiel Richter
Levin
,
Nurit
Adiram-Filiba
and
Eyal
Golub
*
,
Adiel Richter
Levin
,
Nurit
Adiram-Filiba
and
Eyal
Golub
*
Department of Chemistry, Faculty of Exact Sciences and Institute of Nanotechnology and Advanced Materials, Bar Ilan University, 5290002, Israel. E-mail: Eyal.Golub@biu.ac.il
First published on 11th June 2024
Abstract
The microperoxidase-11 hemopeptide exhibits configuration-dependent selectivity for guanine-quadruplexes by specifically uncaging c-MYC guanine-quadruplexes from a duplex DNA.
The secondary structures of genomic DNA have been shown to profoundly influence cellular events and the modulation thereof. While double-stranded B-form DNA (dsDNA) is the predominant and best-studied form, additional non-canonical structures have been identified in past decades, attracting growing research interest.1,2 These include guanine-quadruplex (GQ), which are globular guanine-rich secondary DNA or RNA structures. The formation of Hoogsteen-type H-bonds between guanine nucleotides leads to their assembly into planar tetrads termed guanine quartets (G-tetrad), that subsequently stack one on top of each other through π–π interactions to form a layered structure with various topologies.3,4 Computational surveys of the human genome have identified over 700
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 putative sites of DNA GQ sequences in the genome,5 especially enriched at promoters, flanking the transcription start site, and at the 5′-end of the first intron. Also, the formation of GQs has been suggested to regulate various cellular processes including transcription, translation, pre-RNA splicing, and telomere elongation and they have been widely implemented for biotechnological purposes.6–9 To underpin the specific roles of GQ sequences and visualize them in cells an array of GQ-specific ligands have been developed, exhibiting impressive preference over dsDNA.10–12 However, often the strong affinities of the ligands come at the expense of selectivity between the multitude of sequences and topologies of the GQ sites.13 This originates from the fact that most ligands target the aromatic planar core of G-tetrad which is ubiquitous to all GQ sequences. While several ligands were fitted with auxiliary functional groups that can interact with flanking nucleotides and grooves, there is still a necessity to improve ligand selectivity. Interestingly, the configuration of the GQ target as an additional source of specificity has been scarcely explored. The GQ structure is highly flexible and exists in dynamic equilibria inside cells, transitioning between the GQ and dsDNA forms through various intermediates, Fig. S1 (ESI†). However, the majority of GQ-targeting ligands focus on single-stranded GQs (ssGQs), whereas the influence of flanking nucleotides and configuration of the GQ sequence on ligand binding has remained largely unexplored.14 As such, developing ligands that can either bind, let alone selectively target GQ sequences in the dsDNA configuration will be valuable as an additional mechanism for ligand selectivity. From a design point-of-view, the structure of a ligand that can efficiently bind GQs embedded within dsDNA should contain two components: (i) a dsDNA binding module that will lead to a non-selective binding towards the dsDNA and subsequently concentrating it close to the GQ, and (ii) a GQ-binding module that will enable its binding to the target. While polyaromatic molecules had proven to be tight-binders towards the core of GQ, i.e. the G-tetrads, peptides have shown affinities towards either G-tetrads, grooves, or flanking loops as well as dsDNA,15 and would, thus, complement the polyaromatic module. As such, we sought to employ peptide-linked GQ ligands to target dsDNA-GQ constructs.
000 putative sites of DNA GQ sequences in the genome,5 especially enriched at promoters, flanking the transcription start site, and at the 5′-end of the first intron. Also, the formation of GQs has been suggested to regulate various cellular processes including transcription, translation, pre-RNA splicing, and telomere elongation and they have been widely implemented for biotechnological purposes.6–9 To underpin the specific roles of GQ sequences and visualize them in cells an array of GQ-specific ligands have been developed, exhibiting impressive preference over dsDNA.10–12 However, often the strong affinities of the ligands come at the expense of selectivity between the multitude of sequences and topologies of the GQ sites.13 This originates from the fact that most ligands target the aromatic planar core of G-tetrad which is ubiquitous to all GQ sequences. While several ligands were fitted with auxiliary functional groups that can interact with flanking nucleotides and grooves, there is still a necessity to improve ligand selectivity. Interestingly, the configuration of the GQ target as an additional source of specificity has been scarcely explored. The GQ structure is highly flexible and exists in dynamic equilibria inside cells, transitioning between the GQ and dsDNA forms through various intermediates, Fig. S1 (ESI†). However, the majority of GQ-targeting ligands focus on single-stranded GQs (ssGQs), whereas the influence of flanking nucleotides and configuration of the GQ sequence on ligand binding has remained largely unexplored.14 As such, developing ligands that can either bind, let alone selectively target GQ sequences in the dsDNA configuration will be valuable as an additional mechanism for ligand selectivity. From a design point-of-view, the structure of a ligand that can efficiently bind GQs embedded within dsDNA should contain two components: (i) a dsDNA binding module that will lead to a non-selective binding towards the dsDNA and subsequently concentrating it close to the GQ, and (ii) a GQ-binding module that will enable its binding to the target. While polyaromatic molecules had proven to be tight-binders towards the core of GQ, i.e. the G-tetrads, peptides have shown affinities towards either G-tetrads, grooves, or flanking loops as well as dsDNA,15 and would, thus, complement the polyaromatic module. As such, we sought to employ peptide-linked GQ ligands to target dsDNA-GQ constructs.
Microperocxidase 11 (MP-11) is a product of proteolytic digestion of c-type cytochrome from an equine heart that has been extensively employed as a model for peroxidase enzymes, Fig. 1.16,17 This metallopeptide contains an aromatic heme moiety covalently bound to an 11-mer peptide chain by two cysteines. The proximal site of the heme is coordinated with a histidine residue and the distal site is exposed to solution. A previous study demonstrated the ability of MP-11 to bind to GQ through the heme moiety that interacts in π–π stacking through the distal site.18 As such, the heme, a known GQ ligand,19 serves as a docking unit to the GQs, while the flexible peptide chain engages in non-covalent interactions with the GQ structure. Importantly, the positive charge of the peptide chain renders it an excellent candidate for interacting and binding to GQs embedded in the dsDNA form. However, in the original report, the free amines of the peptide in the Nterm and 13 K residues were acetylated (AcMP-11) to prevent ligand aggregation, thereby neutralizing residues essential for dsDNA binding. Accordingly, the binding of the native MP-11 towards GQs and GQs embedded within dsDNA has not been explored.
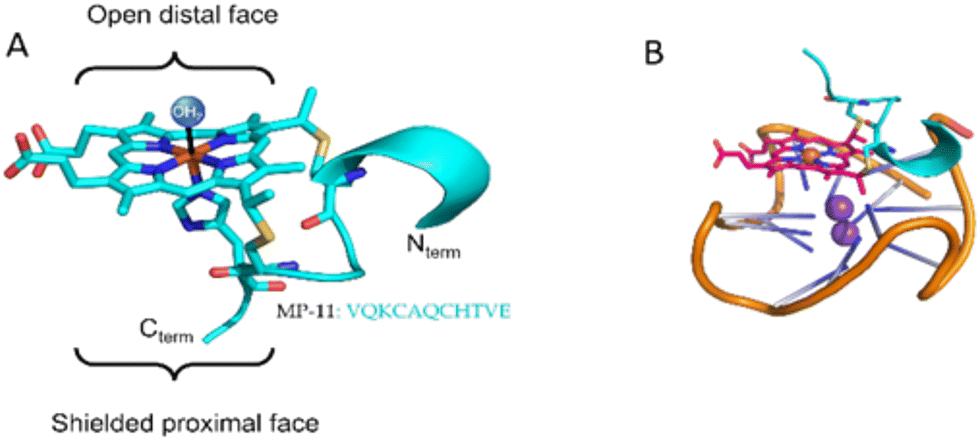 | ||
| Fig. 1 (A) Structure of MP-11 based on the crystal structure of cytochrome c (PDB: 1CGO). (B) Cartoon representation of the complex between c-MYC GQ and MP-11. | ||
We report here on the synergistic interplay between the heme moiety and the peptide chain of MP-11 in binding a representative library of GQ sequences. We find that this set of ssGQs binds MP-11 to yield MP-11/GQ complexes exhibiting variable dissociation constants. Moreover, we show that MP-11 extracts only the c-MYC GQ from a duplex assembly to form the MP-11/c-MYC while none of the other GQs in the duplex form are extracted. That is, c-MYC GQ reveals a selective binding pattern, as compared to other GQ constituents. We attribute this to a non-specific interaction of the peptide chain of MP-11 with the GQ-flanking nucleotides, that concentrate it near the GQ target, thus allowing it to bind via the heme moiety. These interactions are also attenuated by pH, as in mildly acidic conditions the binding of the MP-11 are intensified due to both reduced aggregation of the ligand and co-folding of the GQ with a complementary secondary DNA structure, i.e., the i-motif.
We first investigated the interactions between the peptide and a set of GQs using UV-Vis spectroscopy to determine if the MP-11 binds GQs similarly to AcMP-11. MP-11 is known to partially homooligomerize via nitrogenous ligands (Nterm and 13 K)20 which leads to a low-spin species with characteristic optical absorption bands, Fig. 2A, that can alter significantly upon ligand binding.17 MP-11 exhibited binding to c-MYC GQ at pH = 7.5 as a spectral shift of the Q bands of MP-11 was observed, with the appearance of a charge-transfer band at 625 nm, along with both narrowing of the Soret band and a blue shift from 404 nm to 401 nm. These phenomena represent both the monomerization of MP-11 as well as the transition of the heme moiety from a low-spin to a high-spin state, consistent with previous reports, by exchanging the N-donor ligand from another MP-11 molecule with a water molecule that is associated with the GQ structure.20 To learn about the binding mode of MP-11 with c-MYC GQ we monitored the peroxidase activity of MP-11 as a function of the GQ concentration. The binding of MP-11 to the GQ via stacking of the distal site on the G-tetrad would result in inhibition of the inherent peroxidase activity of the metallopeptide. To verify this binding mode we measured the MP-11-catalyzed oxidation of 2,2′-Azino-di-(3-ethylbenzothiazoline)-6-sulfonic acid (ABTS2−) with H2O2, Fig. 2B. Indeed, the peroxidase activity of MP-11 was inhibited in the presence of GQ in a dose-dependent manner. This implied that the distal site of MP-11 is blocked due to the coordination of the GQ ligand, thus preventing the interaction of H2O2 with the catalytic iron center. This indicates that the heme moiety binds to the external G-tetrad of the GQ via the distal site, leading to the observed spectral blue shift. Moreover, circular dichroism (CD) studies probing the interactions of MP-11 with c-MYC GQ, Fig. 2C, further support a specific binding mode of MP-11 to the c-MYC GQ. As c-MYC GQ folds into a parallel motif, Fig. 2C, curve a, and as hemin was reported to prefer to bind to parallel GQ structures, it was reasonable to assume that effective binding of MP-11 to the c-MYC GQ will proceed. Indeed, Fig. 2C, curve b, demonstrates that the CD band at 265 nm of the parallel c-MYC remained unchanged upon MP-11 binding, supporting the superior association of MP-11 to the c-MYC framework.
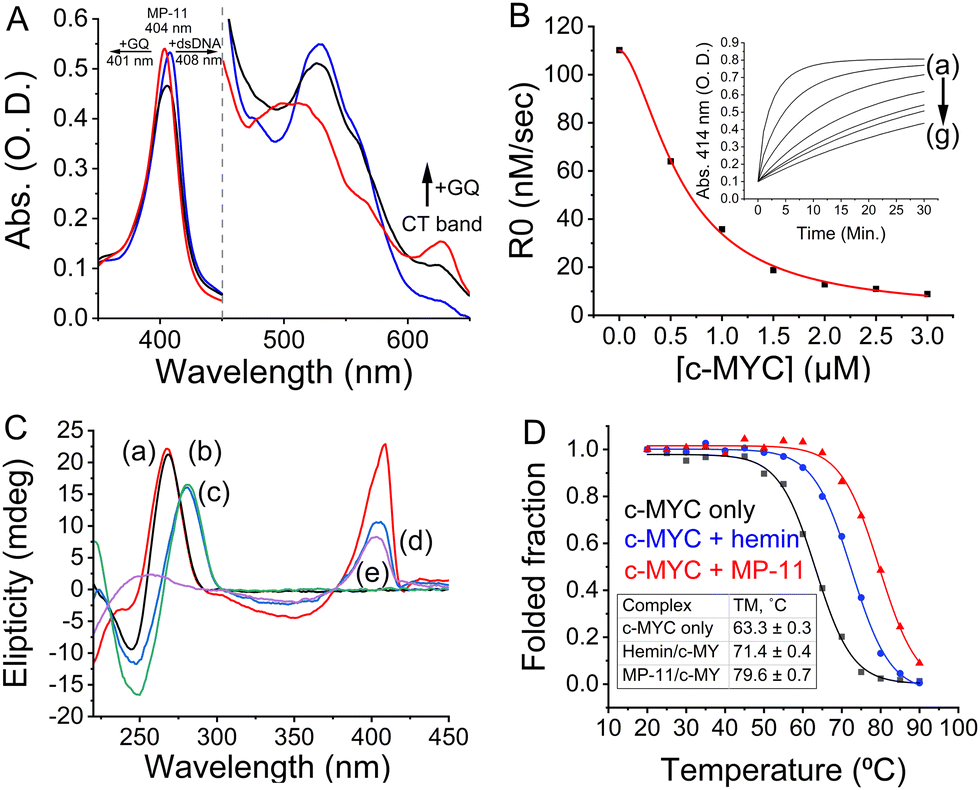 | ||
| Fig. 2 (A) UV-Vis spectra of MP-11 in the absence of DNA (black curve) or in the presence of c-MYC GQ (red curve) or dsDNA (blue curve). (B) The initial rate for the MP-11-catalyzed H2O2-mediated oxidation of ABTS2− in the presence of variable concentrations of c-MYC. Inset: Time-dependent spectral changes at λ = 414 nm corresponding to the oxidation of ABTS2− in the presence of variable concentrations of c-MYC (a) 0 μM, (b) 0.5 μM, (c) 1.0 μM, (d) 1.5 μM, (e) 2.0 μM, (f) 2.5 μM and (g) 3.0 μM. (C) CD spectra corresponding to (a) c-MYC, (b) c-MYC + MP-11, (c) dsDNA, (d) dsDNA + MP-11, and (e) MP-11. (D) Normalized CD melting curves of c-MYC alone (black curve) or in the presence of hemin (blue curve) or MP-11 (red curve). | ||
The optical properties of MP-11 proved to be instrumental in determining the binding mode of MP-11 to DNA targets, Fig. S2 (ESI†). When MP-11 interacted with dsDNA we observed a red shift of the Soret band to 408 nm and the disappearance of the CT band, Fig. S2B (ESI†). As the parent hemin cofactor doesn’t interact with dsDNA structures,19 we attributed the spectral change to the electrostatic binding between the peptide chain of MP-11 and the DNA backbone, resulting in a non-specific binding mode which, in turn, promoted the oligomerization of DNA-bound MP-11 that eliminated the CT band. This non-specific binding mode was also observed for the Htel GQ that forms an anti-parallel topology, Fig. S2C (ESI†). Also, the spectroscopic data from the interactions MP-11 with other parallel GQs of c-KIT and KRAS reveals a red shift of the Soret band accompanied by minute CT bands at 625 nm for the two complexes, Fig. S2D and F (ESI†). This suggests that a partial specific association of the MP-11 constituents in the resulting structures cannot be excluded. On the other hand, the hybrid 532 sequence exhibited a specific binding mode to MP-11. Interestingly, our ability to distinguish between the binding modes was abolished when we employed the AcMP-11 which lacks free amines. The interaction of AcMP-11 with DNA was accompanied by a shift of the Soret band from 396 nm to 401 nm with both sequences, Fig. S3 (ESI†). Thus, the changes in the absorbance features of MP-11 upon binding to the GQs provide a useful tool for identifying the binding modes in the resulting complexes. While a shift of the Soret band to λ = 408 nm implies an electrostatic, non-specific binding, the Soret band shift to λ = 401 nm indicates an ordered π-stacked configuration between MP-11 and the GQ framework (demonstrated for c-MYC and 532 GQs).
We next studied the peptide chain's role in stabilizing the MP-11/GQ supramolecular complex. The association of the parent hemin to GQs allowed us to isolate the influence of the peptide chain when forming the complex with the c-MYC GQ. Accordingly, we measured the melting temperature of the complexes by CD spectroscopy, Fig. 2D. The MP-11/c-MYC GQ complex exhibited enhanced thermal stability as compared with the hemin complex with ΔTM = 8.2 °C and both of them were stabilized as compared with the bare c-MYC sequence with ΔTM,MP-11 = 16.3 °C and ΔTM,hemin = 8.1 °C. On the contrary, no change in the TM was observed when Htel sequence was employed, Fig. S4 (ESI†). As such, the peptide chain is instrumental in binding and stabilizing the formed complex. To account for the chaperone-like role of the peptide chain, the c-MYC sequence was folded in the presence of Na+-containing buffer where it mainly folded into an anti-parallel topology with a small portion of the oligonucleotide remaining unfolded, Fig. S5 (ESI†). The addition of MP-11 not only induced the folding of c-MYC GQ into a parallel topology but also enabled the conversion of antiparallel c-MYC into a parallel topology that binds to MP-11. Given hemin's chaperone-like activity, we inferred that the specific binding of MP-11 to c-MYC is primarily mediated by the heme prosthetic group, which targets the G-tetrad, with the peptide chain binding to the flanking residues.
We, then, investigated the binding of the MP-11 ligand towards GQ sequences that were embedded within a dsDNA structure, termed dsDNA:GQ, and are thus presumably unavailable to bind MP-11, Fig. 3A. The GQ sequences were flanked on both ends with 19 bps and were added with the complementary sequence, termed ssDNA:GQ and ssDNA:GQcomp, respectively. At pH = 7.5 no specific binding mode was observed as the Soret peak shifted to λ = 408 nm, similarly to dsDNA, and the CT band vanished, Fig. S6B–F (ESI†). An exception was observed with dsDNA:c-MYC where a complex formation resulted in a single peak at 405 nm and an enhanced CT band at 625 nm, however, without any clear isosbestic point, Fig. S6A (ESI†). As the Soret band shifts to an intermediate value between the π-stacked specific binding mode of λ = 401 nm and the non-specific electrostatic binding mode of λ = 408 nm, we conclude that MP-11 binds to the duplex and partially extracts and binds the c-MYC GQ. To enable a clearer complex characterization, we performed the binding measurements at pH = 5.5. Under mildly acidic conditions the Nterm of MP-11 is positively charged, and coordinate poorly to the iron ion of an additional MP-11 ligand, thus inhibiting the aggregation of MP-11. Indeed, the Soret band of MP-11 was shifted to 398 nm with an observable CT band, implying monomerization, Fig. S7A (ESI†). Similarly to pH = 7.5, the interactions between MP-11 and the oligonucleotides resulted in a mode-specific spectrum. The interaction with dsDNA led to a decrease in the intensity of the Soret band and the disappearance of the CT band, Fig. S7B (ESI†). Similar behavior was observed with the antiparallel Htel GQ, implying that the changes were correlated to non-specific binding of MP-11 to DNA, Fig. S7C (ESI†). On the other hand, MP-11 exhibited pH-dependent binding to all parallel GQs including c-MYC, c-KIT, and KRAS as well as the hybrid 532 GQ. The interaction was accompanied by a two-phase binding mode, Fig. S7A and D–F (ESI†). At lower concentrations of the GQ, the intensity of the Soret band, λ = 398 nm decreased, similarly to dsDNA. In the second phase, the increase in the concentration of the ligand led to a shift of the Soret band to 403 nm followed by a concentration-dependent increase in the intensity, with an isosbestic point at 417 nm for all sequences. This implied that at lower concentrations MP-11 was binding to parallel GQs through non-specific interactions, while an increase in the concentration of the ligand resulted in a specific stacking of the heme moiety of the MP-11 ligand on the G-tetrad. As this binding mechanism was identical to all parallel GQ sequences as well, it implied a specific binding mode. c-MYC exhibited the highest affinity towards MP-11 with an apparent dissociation constant corresponding to Kd = 6.3 ± 0.8 μM, Fig. S7G (ESI†). When MP-11 interacted with dsDNA:c-MYC a binding profile similar to free c-MYC GQ was observed, Fig. 3B. To verify that MP-11 was binding to the caged GQ we introduced a G17A mutation, thus preventing the folding of the GQ. The mutated sequence did not interact specifically with the ligand as a binding profile similar to dsDNA was observed, Fig. 3C. Thus, we concluded that MP-11 was binding to the c-MYC GQ by extracting it from the duplex dsDNA:c-MYC system. Importantly, all other dsDNA:GQ sequences exhibited spectral binding features identical to that of dsDNA, implying they were binding electrostatically to the DNA, and the GQ sequences remained caged, Fig. S8 (ESI†).
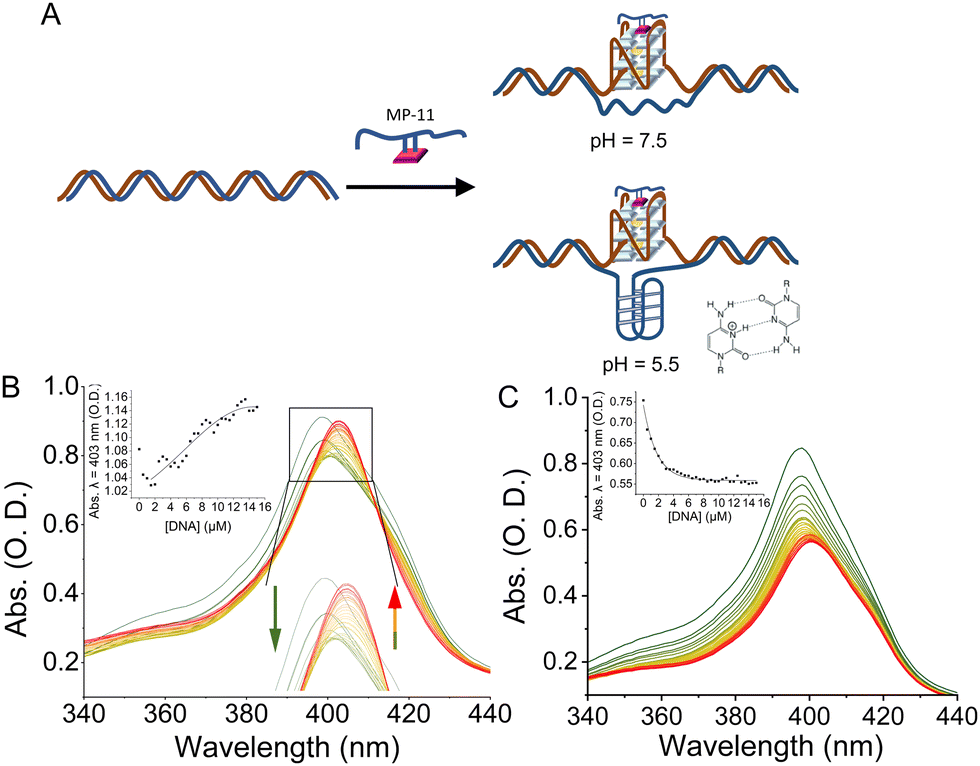 | ||
| Fig. 3 (A) Cartoon representation of the dsDNA:GQ system where a GQ sequence is embedded in the center of the dsDNA. The addition of the native MP-11 ligand leads to the extraction of the GQ sequence from the dsDNA and the formation of a MP- 11/GQ complex. At acidic pHs the binding event is accompanied by the formation of an i-motif structure. (B) and (C) Changes in the UV-Vis spectra of the Soret band of MP-11 as a function of increasing concentration of: (B) dsDNA:c-MYC and (C) dsDNA:G17Ac-MYC with MP-11. Insets: Titration curves monitoring the changes in the intensity at λ = 403 nm. Reactions were performed at pH = 5.5, 50 mM KCl. | ||
To explain this observation, we first account for the electrostatic charge of the peptide. The two propionic acids of the heme moiety have a pKa of 6.1, implying that at pH = 5.5 they will be mostly protonated, and thus, the repulsive electrostatic interactions between MP-11 and the double-stranded negatively charged duplex framework would be depleted. Consequently, the association of MP-11 to the duplex should be facilitated. Accordingly, in this binding mechanism, MP-11 would first bind non-specifically to dsDNA, thus increasing the concentration of the ligand in close proximity to the GQ target until a critical concentration is reached, Fig. 3B. To test this, we explored the interaction of MP-11 ligand with a series of ssDNA:c-MYC sequences that contained an increasing number of flanking nucleotides on both ends of the GQ as non-specific binding sites, Fig. S9 (ESI†), corresponding to an addition of 5, 10 and 20 nucleotides. These DNA constructs would allow us to determine the influence of the addition of nucleotides in close vicinity to the GQ sequence. Importantly, we systematically added the nucleotides at both the 5′ and the 3′ ends, implying that the close vicinity of the GQ site remained unchanged and thus unperturbed in each of the sequences. ssDNA:c-MYC exhibited enhanced binding to MP-11 as compared with c-MYC with Kd = 4.3 ± 0.4 μM. Also, we observed that the affinity of the ligand increased with an increasing number of nucleotides with Kd(+5) = 3.8 ± 0.4 μM. Interestingly, a further increase in the number of flanking nucleotides led to a decrease in affinity with Kd(+10) = 7.2 ± 0.4 μM, and negligible binding was observed for ssDNA:c-MYC(+20). This is explained by the fact that the added nucleotides as non-specific binding sites also compete with the GQ for the MP-11 ligand, and at high numbers can efficiently sequester it. Similarly, in the dsDNA:c-MYC system the flanking residues enrich the hybridized system in MP-11 ligands, Fig. 4, although it binds with lower affinity with Kd = 6.9 ± 1.1 μM, Fig. 4, curve a. When we explored the binding of MP-11 to dsDNA:c-MYC with an increasing number of flanking residues an opposite trend was observed to that of ssDNA:c-MYC. The binding affinity of the ligand towards the GQ decreased with an increasing number of nucleotides, where no binding was observed for +20 nucleotides, Fig. 4, curves b–d. This implied that increasing the number of negatively charged phosphate sites in the duplex system outcompeted the molarity effect of MP-11.
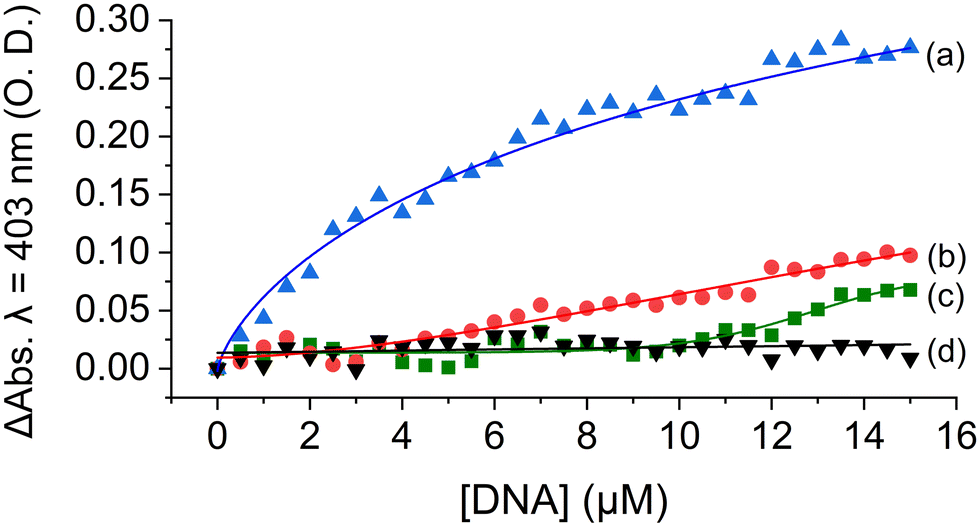 | ||
| Fig. 4 Titration curves of MP-11 at pH = 5.5 in the presence of dsDNA:c-myc encoding for an increasing number of flanking nucleotides: (a) 0 (original sequence), (b) +5, (c) +10 and (d) +20. The curves correspond to the net changes in the absorption at 403 nm compared with dsDNA. | ||
Finally, an important driving force for the extraction of GQ from dsDNA is the folding of the C-rich complementary strand of the GQ into a globular structure, termed i-motif at mildly acidic conditions. This folding process was shown to be mutually exclusive with GQ formation and assists in the folding-out of both structures from the dsDNA form.21
This work explored another layer of selectivity of ligands for GQs. Native MP-11 exhibited selective binding to parallel and hybrid GQs through a synergistic interplay between its heme and peptide components. Also, a configurational selectivity was observed as MP-11 was selectively binding to c-MYC GQs caged in a duplex form over other GQs. This data suggests that the configuration of GQs should also be considered in ligand design.
Conflicts of interest
There are no conflicts to declare.References
- S. Burge, et al. , Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS PubMed.
- J. Spiegel, et al. , Trends Chem., 2020, 2, 123–136 CrossRef CAS PubMed.
- D. Sen and W. Gilbert, Nature, 1988, 334, 364–366 CrossRef CAS PubMed.
- C. J. Lech, et al. , Nucleic Acids Res., 2013, 41, 2034–2046 CrossRef CAS PubMed.
- V. S. Chambers, et al. , Nat. Biotechnol., 2015, 33, 877–881 CrossRef CAS PubMed.
- P. Rawal, et al. , Genome Res., 2006, 16, 644–655 CrossRef CAS PubMed.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed.
- M. P. O’Hagan, et al. , Eur. J. Org. Chem., 2019, 4995–5017 CrossRef.
- J. Dong, et al. , Chem. Soc. Rev., 2022, 51, 7631–7661 RSC.
- A. De Cian, et al. , Biochimie, 2008, 90, 131–155 CrossRef CAS PubMed.
- D. Monchaud and M.-P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627–636 RSC.
- J. Zegers, M. Peters and B. Albada, J. Biol. Inorg. Chem., 2023, 28, 117–138 CrossRef CAS PubMed.
- M. Zuffo, et al. , Nucleic Acids Res., 2018, 46, e115 CrossRef PubMed.
- J. Chen, et al. , Nucleic Acids Res., 2021, 49, 9548–9559 CrossRef CAS PubMed.
- B. Heddi, et al. , Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9608–9613 CrossRef CAS PubMed.
- D. A. Baldwin, et al. , J. Inorg. Biochem., 1987, 30, 203–217 CrossRef CAS PubMed.
- H. M. Marques, Dalton Trans., 2007, 4371–4385 RSC.
- J. Liu, et al. , Chem. Commun., 2023, 59, 7811–7814 RSC.
- P. Travascio, et al. , Chem. Biol., 1998, 5, 505–517 CrossRef CAS PubMed.
- H. M. Marques and C. B. Perry, J. Inorg. Biochem., 1999, 75, 281–291 CrossRef CAS PubMed.
- Y. Cui, et al. , Biochemistry, 2016, 55, 2291–2299 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01389a |
| This journal is © The Royal Society of Chemistry 2024 |