
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold carbonyl cations and beyond: homoleptic gold(I) complexes with P4 and P4S3 and the half-sandwich cation [Au(C6H6)(CO)]+†
Manuel
Schmitt
 ab,
Sarah
Nestle
a,
Valentin
Radtke
a and
Ingo
Krossing
*a
ab,
Sarah
Nestle
a,
Valentin
Radtke
a and
Ingo
Krossing
*a
aAlbert-Ludwigs University Freiburg, Albertstr. 21, Freiburg 79104, Germany. E-mail: krossing@uni-freiburg.de
bUniversität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
First published on 24th April 2024
Abstract
Oxidation of Au0 with the synergistic Ag+/0.5 I2 system in the commercial organic solvent 1,2,3,4-tetrafluorobenzene led to the perfluoroalkoxyaluminate salt of the [Au(CO)2]+ cation known from superacid chemistry. This [Au(CO)2]+ salt proved to be an excellent ‘naked’ Au+-synthon yielding complex salts with [Au(η2-P4)2]+, [Au(η1-P4S3)2]+ and half-sandwich [Au(η2-C6H6)(CO)]+ cation.
The chemistry of gold is dominated by relativistic effects, which cause a stabilization of the 6s orbital and an expansion of the 5d orbitals.1,2 As a result, relativistic effects increase the ionization energy, electronegativity and atomization energy of gold compared to the lighter coinage metals copper and silver.2–4 Hence, gold compounds mainly exhibit the oxidation states +I and +III with linear or square-planar coordination.1 A simple Au(I) prototype is the linear gold dicarbonyl cation [Au(CO)2]+, first isolated by Aubke and Wilner during the attempted protonation of CO in the superacid HSO3F–Au(SO3F)3.5 In general, such transition metal carbonyl cations (TMCCs) are suitable MI precursors, since the M–CO bonds in TMCCs are rather weak compared to neutral or anionic carbonyl complexes. These weakened M–CO bonds are caused by reduced π-back donation.6–8 This synthetic value of TMCCs was already demonstrated with the synthesis of [M(arene)(CO)4]+ (M = Nb, Ta; arene = C6H3Me3, C6H6, o-dfb = 1,2-F2C6H4)9,10 and [Ni(arene)n(CO)4−2n]+ (arene = C6H6, o-dfb, n = 1, 2)11,12 from [M(CO)7][Al(ORF)4] (M = Nb, Ta) and [Ni(CO)4][F{Al(ORF)3}2] (RF = C(CF3)3), respectively.
Hitherto, the [Au(CO)2]+ cation was investigated in the gas phase by mass spectrometry13 and isolated as mixed [SbF6]−/[Sb2F11]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14–16 or [UF6]−17 salt, yielding vibrational, NMR, and scXRD data. All these reactions were performed in superacidic media. This hampers the general synthetic access to such salts and also the investigation of their follow-up chemistry. An earlier attempt to obtain [Au(CO)2]+ in organic media from AuCl and Ag[Al(ORF)4] in CO atmosphere only yielded the chloride-bridged A-frame salt [Cl{Au(CO)}2]+.18
14–16 or [UF6]−17 salt, yielding vibrational, NMR, and scXRD data. All these reactions were performed in superacidic media. This hampers the general synthetic access to such salts and also the investigation of their follow-up chemistry. An earlier attempt to obtain [Au(CO)2]+ in organic media from AuCl and Ag[Al(ORF)4] in CO atmosphere only yielded the chloride-bridged A-frame salt [Cl{Au(CO)}2]+.18
Here, we present a simple synthesis in the commercial organic solvent 1,2,3,4-tetrafluorobenzene (4FB) that is very weakly coordinating, yet polar (εr = 12.6).19 [Au(CO)2][F{Al(ORF)3}2] 1 was synthesized by oxidation of gold powder with the synergistic Ag+/0.5 I2 system (eqn (1)). Colorless crystals of 1 were obtained by vapor diffusion of n-pentane into a 4FB solution in 70% crystalline yield.
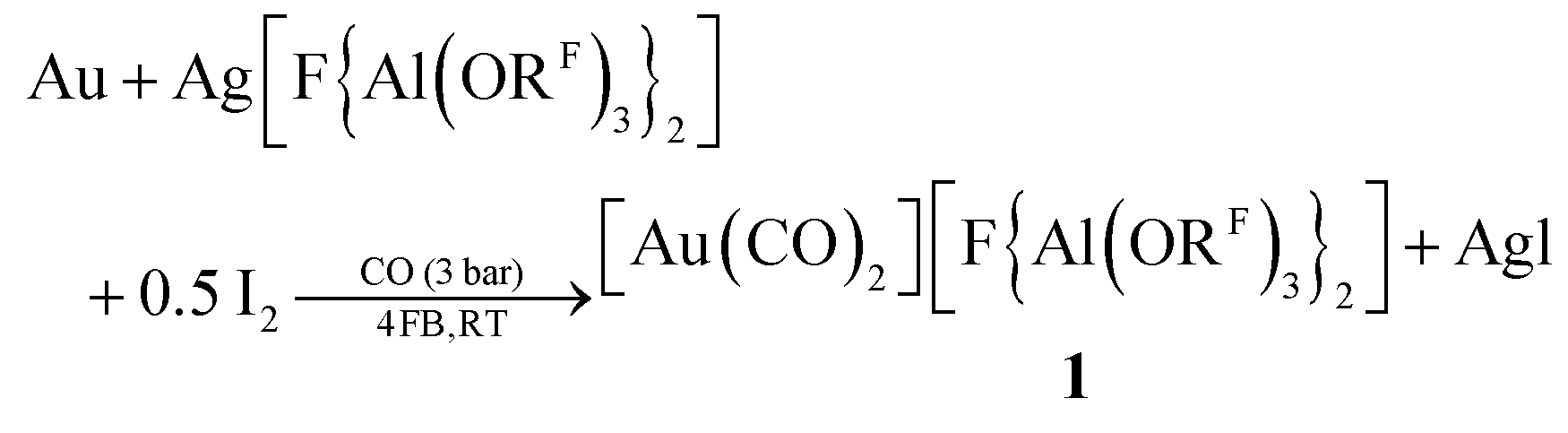 | (1) |
 | ||
Fig. 1 Molecular structure of one [Au(CO)2]+ cation in the crystal structure of 1 (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , R1 = 2.9%, wR2 = 8.1%, thermal ellipsoids shown at the 50% probability level), together with the carbonyl region of the experimental and DFT-simulated IR spectrum of 1 (@B3LYP(D3BJ)/def2-TZVPP level, without scaling factor). , R1 = 2.9%, wR2 = 8.1%, thermal ellipsoids shown at the 50% probability level), together with the carbonyl region of the experimental and DFT-simulated IR spectrum of 1 (@B3LYP(D3BJ)/def2-TZVPP level, without scaling factor). | ||
To prove the synthetic value of 1, we investigated the reaction with the inorganic cages P4 and P4S3 (Scheme 1). In both cases, a complete consumption of the normally insoluble P4 and P4S3 was observed within 1 to 2 h. By vapor diffusion of n-pentane into a 4FB solution of the products, colorless crystals of [Au(P4)2][F{Al(ORF)3}2] 2 (crystalline yield: 68%) and [Au(P4S3)2][F{Al(ORF)3}2] 3 (crystalline yield: 61%) were obtained. Compounds 1, 2 and 3 are stable at RT and were fully characterized by means of vibrational and NMR spectroscopy, as well as single-crystal XRD. Bulk purity was proven with powder XRD (see ESI†, Section S8).
 | ||
| Scheme 1 Investigated reactions of 1 in 4FB with the inorganic cages P4 and PsS3 and the organic molecules acetylene and benzene. | ||
Hence, 1 suites to synthesize the known [Au(P4)2]+ cation, earlier obtained as [GaCl4]− salt by reaction of AuCl with GaCl3 and P4.21 The molecular structure of the [Au(P4)2]+ cation in 2 is displayed in Fig. 2a. Similar to earlier M(P4)-complexes, the P4 molecule coordinates in an η2 fashion21–24 to M = Cu, Ag, Au. The average Au–P distance amounts to 2.4478(8) Å in 2, compared to 2.4430(8) Å in [Au(P4)2][GaCl4].21 It is noteworthy that the P1-P2–P2′-P1′ torsion angle between the two P4 molecules is highly sensitive towards the environment. In gas phase calculations (@B3LYP(D3BJ)/def2-TZVPP and r2scan-3c) and in the [Au(P4)2][GaCl4] salt, this torsion angle is close to 0°, resulting in an almost D2h symmetric [Au(P4)2]+ molecule. In [Ag(P4)2][Al(ORF)4] and [Cu(P4)2][Al(ORF)4] salts, this torsion angle ranges between 4.16(11)° and 13.84(10)°.23,24 In contrast, this torsion angle amounts to 43.76(5)° in [Ag(P4)2][F{Al(ORF)3}2]22 and 37.36(3)° in 2, which reduces the symmetry of the [Au(P4)2]+ cation to D2. The differences in the torsion angles are likely caused by crystal-packing effects and the interactions with the different anions [GaCl4]−, [Al(ORF)4]−, and [F{Al(ORF)3}2]−. In the gas phase, the energy difference ΔE between two conformers of the [Au(P4)2]+ cation with a torsion angle of 0° and 90° amounts to only 0.8 kJ mol−1 at the CCSD(T)/def2-QZVPP//r2scan-3c level of theory (ESI,† Fig. S37).
 | ||
| Fig. 2 (a) Top and side view of the [Au(P4)2]+ cation of the crystal structure of 2 (C2/c, R1 = 1.9%, wR2 = 4.4%). (b) Top and side view of the majority part (86%) of the [Au(P4S3)2]+ cation in 3 (P, R1 = 4.4%, wR2 = 10.9%). (c) Top and side view of the minority part (14%) of the [Au(P4S3)2]+ cation in 3. Thermal ellipsoids are shown at the 50% probability level. | ||
The Raman spectrum of 2 agrees very well with the calculated spectrum at the B3LYP(D3BJ)/def2-TZVPP level of theory (Fig. S22, ESI†). The symmetric breathing mode of the P4 cages in 2 at 593 cm−1 is red-shifted compared to free P4 in the gas phase at 600 cm−1.25 In contrast, the same vibrational modes in [Cu(P4)2][Al(ORF)4] (599 cm−1)23 and [Ag(P4)2][Al(ORF)4] (601 cm−1)23 remain almost unchanged. The increased red-shift illustrates the stronger metal ligand interaction in [Au(P4)2]+ compared to the lighter coinage metals. For a complete assignment of all vibrational modes see ESI,† Section S5. The 31P NMR of 2 shows one singlet resonance at −458.1 ppm, which agrees with the value of [Au(P4)2][GaCl4] at −452 ppm,21 downfield from free P4 at −525 ppm.21
The cyclic voltammograms (CV) of 1 and 2 are shown in the ESI,† Section S7. In both cases, the oxidative and reductive process are coupled and the oxidation is only observed, if a reduction occurred first. However, in both CVs, the anodic and cathodic peak potentials are separated by 0.39 V (1) or 0.80 V (2). This large separation indicates that either the electron transfer reaction is quite slow, or more likely, that the electron transfer is coupled with a chemical reaction. In this case, the neutral [Au(L)2]0 (L = Co, P4) complexes are likely not stable under these conditions at RT and (partial) ligand dissociation occurs. This conclusion is supported by the significantly smaller anodic peak current compared to the corresponding cathodic peak current in the CV of 2 and the considerably reduced bond dissociation Gibbs free energies of neutral [Au(L)2]0 (L = CO: 20 kJ mol−1, L = P4:108 kJ mol−1) compared to [Au(L)2]+ (L = CO: 325 kJ mol−1, L = P4:428 kJ mol−1; @DLPNO-CCSD(T)/def2-QZVPP//r2scan-3c, ESI,† Table S14). This coupled mechanism hinders the determination of a half-wave potential. Therefore, we only report the cathodic peak potentials as the lower limit of the thermodynamic formal potential of the [Au(L)2]+/[Au(L)2]0 couples. The cathodic peak potentials of the compounds amount to 1.40 V (1) and 0.48 V (2), respectively. In accordance with earlier observations,3 the Au+/Au0 potential is very dependent on the environment and is reduced by 0.92 V when exchanging CO for P4.
Next, 1 was used for the generation of the novel AuI complex with the inorganic cage P4S3 (Scheme 1). The [Au(P4S3)2]+ salt 3 was directly accessible. In the molecular structure of 3, the [Au(P4S3)2]+ cation is disordered, and two different coordination isomers were observed. The P4S3 cage coordinates mainly with the apical phosphorus atom to the gold atom (86%, Fig. 2b) and only to a minor extent with one of the basal phosphorus atoms (14%, Fig. 2c). The difference in the Gibbs free energy for the different coordination modes in the gas phase amounts to ΔG = 9.9 kJ mol−1 (@DLPNO-CCSD(T)/def2-QZVPP//r2scan-3c, ESI,† Fig. S38). This coordinative variability of the P4S3 cage was already observed earlier, i.e. for [RuCp(PMe3)2]+,26 [{MeC(CH2PPh2)3}Re(CO)2]+27 or Ag+.28,29 Especially for the coordination of P4S3 to Ag+ with different alkoxyaluminate anions, all three possible η1-binding modes (Papical, Pbasal and S) were observed, but no η2-coordination like in P4 complexes.28,29 The bonding of the P4S3 cage in these complexes is best described as a σ-donation of the P4S3 cage to the metal.27 The frontier orbitals of the P4S3 molecule involved in this interaction are mainly of lone pair character (2 × 3 sulfur and 4 × 1 phosphorus lone pair orbitals).27 Therefore, the bond lengths within the P4S3 cage remain largely unchanged upon coordination to Au+ in 3 (ESI,† Table S8).
The IR and Raman spectra of crystalline 3 agree well with the simulated spectra of the [Au(P4S3)2]+ cation with coordination of the apical phosphorus atom (ESI,† Fig. S25 and S26). However, upon detailed analysis of the spectra, a small band at 427 cm−1 can neither be assigned to the anion nor the [Au(P4S3-apical)2]+ cation and may be caused by the partial coordination of P4S3 with one of the basal phosphorus atoms. In solution, the system is dynamic and the 31P NMR of 3 in 4FB (Fig. S14, ESI†) displays only two signals at +80.7 ppm (Papical) and −122.8 ppm (Pbasal) compared to free P4S3 with signals at +62.6 and −128.7 ppm.30 A similarly dynamic system was reported for solution NMR spectra of [Ag(P4S3)n]+ salts.28,29
Finally, we investigated the reaction of 1 with small organic molecules containing π-bonds (Scheme 1). Hitherto, only few compounds with an unsupported gold arene π-contacts were structurally characterized,31,32 including phosphine,33 NHC34 or CAACs35 as ancillary ligands.
Similarly, AuI acetylene complexes were hitherto only investigated in the gas phase by mass spectrometry and theoretical methods.36 Only [Ag(C2H2)3-4]+37 are known examples of structurally characterized homoleptic transition metal acetylene complexes to date. However, some examples with substituted alkynes like the coinage metal complexes [M(cyclooctyne)3]+ (M = Cu, Ag) and [Au(cyclooctyne)2–3]+ are known in the condensed phase.38 Therefore, we focused on the reaction of 1 with the prototypical molecules benzene and acetylene.
The reactions of 1 in 4FB with an excess of acetylene always resulted in the rapid decomposition of the sample and the precipitation of large amounts of an insoluble black solid of unknown composition, even at low temperatures (ESI,† Fig. S20). Possibly, the electrophilic gold cation in 1 catalyzed the polymerization/functionalization of acetylene, since the activation of alkynes by AuI complexes was reported previously.39
However, addition of benzene to a solution of 1 in 4FB at −35 °C resulted in an immediate gas evolution. By precipitation with hexanes, [Au(C6H6)(CO)][F{Al(ORF)3}2] 4 was obtained as an off-white solid in low yield of 23%. Colorless crystals of 4 were obtained by layering of a 4FB solution with hexanes at −30 °C. Formation of an homoleptic [Au(C6H6)2]+ complex was never observed, even using a large excess of benzene and the application of two freeze–pump–thaw cycles to remove the CO from the equilibrium. DFT calculations revealed that the first CO-arene exchange is favored by ΔRG = −51 kJ mol−1, whereas the second exchange is calculated to be only slightly exergonic by ΔRG = −4 kJ mol−1 (@B3LYP(D3BJ)/def2-TZVPP, Table S16, ESI†) and may explain why we only observed a [Au(C6H6)(CO)]+ complex. 4 is very sensitive to temperature and air and was characterized by IR and NMR spectroscopy as well as single-crystal XRD. At room temperature, 4 decomposes within minutes, as evident from the color change from white to purple, likely caused by the formation of elemental ‘nano-gold’. Elemental gold was also detected by powder XRD (Fig. S36, ESI†), since sample preparation was only possible at RT. However, all signals in the IR and low temperature NMR spectra could be assigned to the target compound 4, indicating the generation of a pure sample at −35 °C.
The [Au(C6H6)(CO)]+ cation in the molecular structure of 4 is disordered over two positions and displays either a η2-coordinating (70%, Fig. 3) or η1-coordinating benzene ring (30%, Fig. S31, ESI†). Gas phase DFT calculations at the B3LYP(D3BJ)/def2-TZVPP level of theory predict the η1-coordination as the minimum structure and the η2-coordination as a transition state (ESI,† Fig. S39). However, both structures are only separated by ΔE = 2.4 kJ mol−1. A similar structural flexibility was observed for [Au(arene)(PR3)]+33 cations and the NiI sandwich complexes [Ni(arene)2]+ (arene = C6H6, o-dfb).11,12 The Au–CO bond lengths of 1.911(9) Å (η2) and 1.884(17) Å (η1) in 4 are shorter than the Au–CO bond in 1 with 1.980(2) Å. This trend is consistent with the stronger electron donating ability of benzene vs. CO and thus, the enhanced Au–CO π-back donation in 4. The stronger Au–CO π-back donation in 4 is also visible in the red-shifted CO stretching frequency of 2200 cm−1 compared to 2219 cm−1 in 1 (Fig. 3, full assignment in ESI,† Section S5). Low temperature NMR spectra at −40 °C revealed only one singlet in the 1H NMR spectrum of 4 in 4FB at 8.00 ppm (ESI,† Fig. S15), downfield of free benzene in 4FB at 7.29 ppm. In the 13C NMR spectrum, the singlet at 127.6 ppm was assigned to the benzene ligand of 4. The observation of only one signal in each NMR spectrum is consistent with the structural flexibility and dynamics of the [Au(C6H6)(CO)]+ cation. Since the experimental efforts to generate a homoleptic [Au(C6H6)2]+ cation were not successful, we analyzed the thermodynamics of essential reactions with several arenes (ESI,† Tables S16 and S17). The Gibbs free energies ΔRG for the formation of [Au(arene)(CO)]+ or [Au(arene)2]+ (arene = C6Me6, Mes, C6H6, o-dfb, 4FB) cation from [Au(CO)2]+ and the corresponding arene were calculated at the B3LYP(D3BJ)/def2-TZVPP level of theory (ESI,† Fig. S40a). As expected, the reaction becomes more exergonic with increasing electron-donating ability of the arene. However, in all cases, the exchange of the first arene is more favored than the second. Since 4 is not stable at RT, we also investigated the potential decomposition reaction, i.e. oxidation of one arene ligand to the corresponding arene radical cation by Au+ (ESI,† Fig. S40b). Interestingly, only the complexes with benzene are predicted to be stable in the series of [Au(arene)(CO)]+ or [Au(arene)2]+ cations. For electron-richer arenes like C6Me6, the stability of the resulting radical cation increases, while for o-dfb and 4FB, the intrinsic Au–arene bond is rather weak. Both trends favor the decomposition of the AuI complexes to Au0(s).
 | ||
| Fig. 3 Majority part (70%) of the [Au(η2-C6H6)(CO)]+ cation in the structure of 4 (P21, R1 = 3.3%, wR2 = 6.7%, thermal ellipsoids shown at 50% probability level), together with experimental and simulated IR spectra of 4 in the carbonyl region. The spectrum was simulated from B3LYP(D3BJ)/def2-TZVPP calculations without a scaling factor. | ||
To conclude, our investigation of weakly bound AuI complexes led to a new synthetic route for the [Au(CO)2]+ cation as perfluoroalkoxyaluminate salt. This room temperature stable compound is accessible in the commercially available, organic solvent 4FB and was used as an excellent synthon for a ‘naked’ Au+ salt, allowing the extension of the coordination chemistry of AuI with weak ligands. With this approach, the homoleptic cations [Au(P4)2]+ and [Au(P4S3)2]+ were obtained. Furthermore, at low temperatures, the half-sandwich complex [Au(C6H6)(CO)]+ was accessible from [Au(CO)2]+ and benzene. This half-sandwich cation represents the first structurally characterized, unsupported gold arene π-contact.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Project numbers 350173756, 431116391). The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the DFG through grant number INST 40/575-1 FUGG (JUSTUS 2 cluster).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- P. Schwerdtfeger, Heteroat. Chem., 2002, 13, 578–584 CrossRef CAS.
- H. Schwarz, Angew. Chem., Int. Ed., 2003, 42, 4442–4454 CrossRef CAS PubMed.
- H. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Chem. Phys., 2005, 311, 151–161 CrossRef CAS.
- H. Willner and F. Aubke, Inorg. Chem., 1990, 29, 2195–2200 CrossRef CAS.
- H. Willner and F. Aubke, Angew. Chem., Int. Ed. Engl., 1997, 36, 2402–2425 CrossRef CAS.
- G. Frenking, J. Organomet. Chem., 2001, 635, 9–23 CrossRef CAS.
- G. Frenking, C. Loschen, A. Krapp, S. Fau and S. H. Strauss, J. Comput. Chem., 2007, 28, 117–126 CrossRef CAS PubMed.
- W. Unkrig, M. Schmitt, D. Kratzert, D. Himmel and I. Krossing, Nat. Chem., 2020, 12, 647–653 CrossRef CAS PubMed.
- W. Unkrig, F. Zhe, R. Tamim, F. Oesten, D. Kratzert and I. Krossing, Chem. – Eur. J., 2021, 27, 758–765 CrossRef CAS PubMed.
- M. Schmitt, M. Mayländer, T. Heizmann, S. Richert, C. Bülow, K. Hirsch, V. Zamudio-Bayer, J. T. Lau and I. Krossing, Angew. Chem., Int. Ed., 2022, 61, e202211555 CrossRef CAS PubMed.
- M. Schmitt, M. Mayländer, V. Radtke, T. Heizmann, S. Richert and I. Krossing, Chem. – Eur. J., 2023, 29, e202301419 CrossRef CAS PubMed.
- J. Velasquez, B. Njegic, M. S. Gordon and M. A. Duncan, J. Phys. Chem. A, 2008, 112, 1907–1913 CrossRef CAS PubMed.
- R. Küster and K. Seppelt, Z. Anorg. Chem., 2000, 626, 236–240 CrossRef.
- C. Wang, S. C. Siu, G. Hwang, F. Aubke, C. Bach, B. Bley, M. Bodenbinder and H. Willner, Can. J. Chem., 1996, 74, 1952–1958 CrossRef CAS.
- H. Willner, J. Schaebs, G. Hwang, F. Mistry, R. Jones, J. Trotter and F. Aubke, J. Am. Chem. Soc., 1992, 114, 8972–8980 CrossRef CAS.
- M. Adelhelm, W. Bacher, E. G. Höhn and E. Jacob, Chem. Ber., 1991, 124, 1559–1561 CrossRef CAS.
- J. Schaefer, A. Kraft, S. Reininger, G. Santiso-Quinones, D. Himmel, N. Trapp, U. Gellrich, B. Breit and I. Krossing, Chem. – Eur. J., 2013, 19, 12468–12485 CrossRef CAS PubMed.
- I. Krossing, C. Armbruster, M. Sellin, M. Seiler, T. Würz, F. Oesten, M. Schmucker, T. Sterbak, J. Fischer, V. Radtke and J. Hunger, Research Square, 2024, preprint DOI:10.21203/rs.3.rs-3921217/v1.
- Q. Xu, Y. Imamura, M. Fujiwara and Y. Souma, J. Org. Chem., 1997, 62, 1594–1598 CrossRef CAS.
- L. C. Forfar, T. J. Clark, M. Green, S. M. Mansell, C. A. Russell, R. A. Sanguramath and J. M. Slattery, Chem. Commun., 2012, 48, 1970–1972 RSC.
- A. Bihlmeier, M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem. – Eur. J., 2004, 10, 5041–5051 CrossRef CAS PubMed.
- G. Santiso-Quiñones, A. Reisinger, J. Slattery and I. Krossing, Chem. Commun., 2007, 5046–5048 RSC.
- I. Krossing, J. Am. Chem. Soc., 2001, 123, 4603–4604 CrossRef CAS PubMed.
- I. R. Beattie, G. A. Ozin and R. O. Perry, J. Chem. Soc. A, 1970, 2071–2074 RSC.
- M. Caporali, F. D. Calvo, C. Bazzicalupi, S. Seniori Costantini and M. Peruzzini, J. Organomet. Chem., 2018, 859, 68–74 CrossRef CAS.
- E. Guidoboni, I. de los Rios, A. Ienco, L. Marvelli, C. Mealli, A. Romerosa, R. Rossi and M. Peruzzini, Inorg. Chem., 2002, 41, 659–668 CrossRef CAS PubMed.
- A. Adolf, M. Gonsior and I. Krossing, J. Am. Chem. Soc., 2002, 124, 7111–7116 CrossRef CAS PubMed.
- I. Raabe, S. Antonijevic and I. Krossing, Chem. – Eur. J., 2007, 13, 7510–7522 CrossRef CAS PubMed.
- P. Weis, I. M. Riddlestone, H. Scherer and I. Krossing, Chem. – Eur. J., 2019, 25, 12159–12168 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2–23 CrossRef CAS.
- R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714–11724 CrossRef CAS PubMed.
- E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455–5459 CrossRef PubMed.
- N. Parvin, B. Mishra, A. George, M. Neralkar, J. Hossain, P. Parameswaran, S. Hotha and S. Khan, Chem. Commun., 2020, 56, 7625–7628 RSC.
- V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569–13573 CrossRef CAS PubMed.
- T. B. Ward, A. D. Brathwaite and M. A. Duncan, Top. Catal., 2018, 61, 49–61 CrossRef CAS.
- A. Reisinger, N. Trapp, I. Krossing, S. Altmannshofer, V. Herz, M. Presnitz and W. Scherer, Angew. Chem., Int. Ed., 2007, 46, 8295–8298 CrossRef CAS PubMed.
- A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940–3943 CrossRef CAS PubMed.
- H. C. Shen, Tetrahedron, 2008, 64, 3885–3903 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2270712–2270715. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01374c |
| This journal is © The Royal Society of Chemistry 2024 |