
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of (1-silyl)allylboronates by KOtBu-catalyzed ring-opening gem-silylborylation of cyclopropenes†
Ikuya
Fujii
ab,
Haruka
Hirata
a,
Hirokazu
Moniwa
a and
Ryo
Shintani
 *ab
*ab
aDivision of Chemistry, Department of Materials Engineering Science, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: shintani.ryo.es@osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
First published on 12th June 2024
Abstract
A KOtBu-catalyzed ring-opening gem-silylborylation of cyclopropenes with silylboronates has been developed for the synthesis of (1-silyl)allylboronates, a useful class of compounds in organic synthesis. The reaction proceeds with high selectivity under mild conditions, and the reaction mechanism has been theoretically investigated using DFT calculations.
Allylic organometallic reagents are highly valued and play important roles in synthetic organic chemistry, particularly due to their broad and versatile reactivity.1 Among them, allylsilanes and allylboranes are distinguished by their facile manipulation and exceptional selectivity in various transformations.1e–i,2 In this regard, (1-silyl)allylboranes, a particular class of allylic gem-dimetalloids possessing both silicon and boron functionalities at the same allylic carbon, are anticipated to serve as useful building blocks for the synthesis of complex molecules.3 The high potential of (1-silyl)allylboranes has been empirically evidenced by the broad scope of transformations through regio-, chemo-, and stereoselective C–C bond forming reactions.3,4
Despite the utility of these compounds, their efficient synthetic methods remain to be further developed. Most of the existing synthetic strategies rely on the use of preformed organosilicon and/or organoboron compounds as exemplified by silylation of allylboranes (Scheme 1a),5a hydroboration of allenylsilanes (Scheme 1b)5b or silylcyclopropenes,5c homologation of alkenylboronic acid derivatives with silylmethylene donors (Scheme 1c),5d,e and cross-coupling of bromoalkenes with diborylsilylmethanes (Scheme 1d).5f In contrast, direct introduction of both silicon and boron substituents into the allylic position has been essentially limited to gem-silylborylation of allylic carbenoids generated from allylic halides with lithium diisopropylamide at a very low temperature (−98 °C; Scheme 1e).3b,5g In this context, herein we describe the development of a new way of synthesizing (1-silyl)allylboronates via a ring-opening gem-silylborylation reaction of cyclopropenes with silylboronates6 in the presence of a catalytic amount of KOtBu (Scheme 1f), representing a rare example of concurrently introducing silicon and boron functional groups to the same allylic carbon under mild conditions.
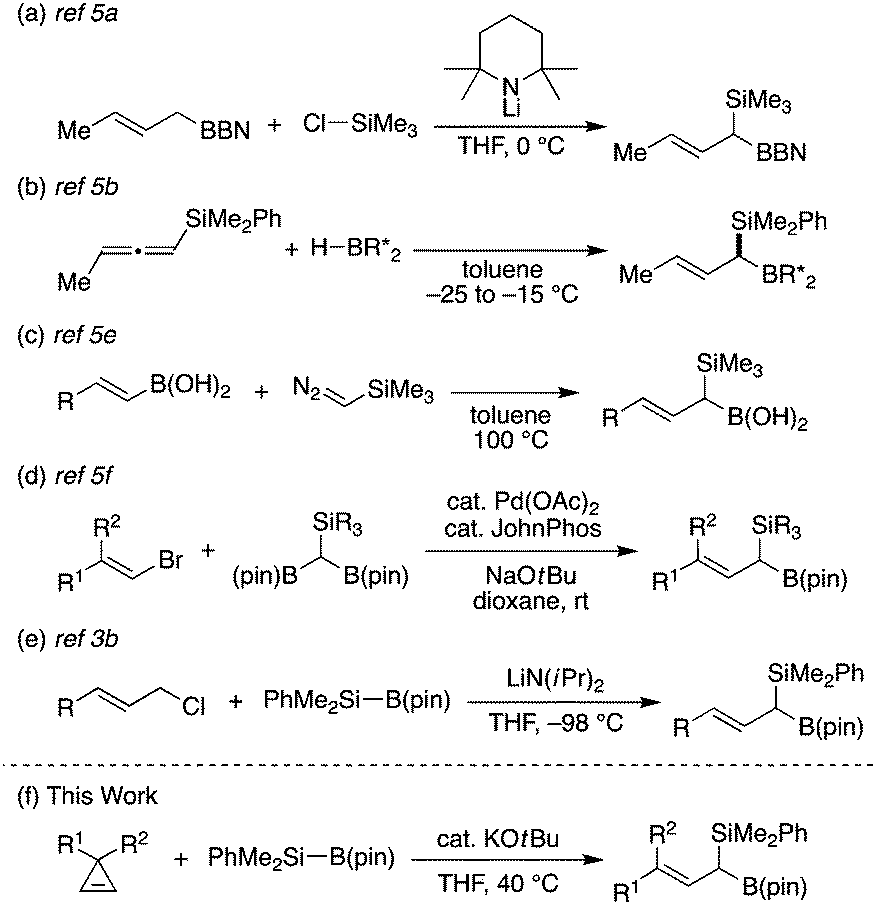 | ||
| Scheme 1 (a)–(e) Reported examples of (1-silyl)allylboron synthesis and (f) ring-opening gem-silylborylation of cyclopropenes (this work). | ||
Initially, we employed 3,3-diphenylcyclopropene (1a) as a model substrate and conducted the reaction with dimethyl(phenyl)silylboronate 2a in THF at 40 °C (Table 1). No significant reaction took place in the absence of a catalyst (entry 1). We then focused on the use of base catalysts to activate 2a as a silicon nucleophile,7 and the reaction in the presence of 10 mol% of KOtBu led to a clean formation of ring-opened (1-(dimethyl(phenyl)silyl)-3,3-diphenylallyl)boronate 3aa in 79% yield (entry 2). This ring-opening gem-silylborylation is in stark contrast to the simple 1,2-silylborylation of styrene derivatives reported by Ito and coworkers under similar reaction conditions.7b A high yield of 3aa was also achieved by using NaOtBu instead of KOtBu (entry 3), but LiOtBu was found to be ineffective presumably due to the lower basicity (entry 4). It is worth noting that the use of a transition-metal alkoxide catalyst such as Cu(OtBu)(IPr)8 led to the formation of a dimer of 1a in the form of 3,3,6,6-tetraphenyltricyclo[3.1.0.02,4]hexane (4) with no formation of 3aa (entry 5).9

Under simple and mild conditions using KOtBu as the catalyst, various 3,3-diarylcyclopropenes 1 could be transformed into the corresponding (1-silyl)allylboronates 3 by the reaction with silylboronate 2a (Table 2). For example, in addition to unsubstituted phenyl group (1a), aryl groups having mildly electron-donating or electron-deficient substituents at the 4-positions (1b–e) were tolerated to give (1-silyl)allylboronates 3aa–ea in moderate to good isolated yields (45–72% yield),10 although highly electron-rich 3,3-di(4-methoxyphenyl)cyclopropene and highly electron-deficient 3,3-di(4-trifluoromethylphenyl)cyclopropene were found to be unreactive (0% and 17% yield, respectively; data not shown in the table). On the other hand, 3-methoxyphenyl (1f) and 2-naphthyl (1g) groups were applicable as substituents on cyclopropenes and products 3fa and 3ga were obtained in 65–68% isolated yields. The structure of 3ga was confirmed by X-ray crystallographic analysis.11 Furthermore, the reaction proceeded well with substrate 1h having one electron-donating 4-methoxyphenyl group and one electron-withdrawing 4-chlorophenyl group, and (1-silyl)allylboronate 3ha was isolated in 56% yield as an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of E/Z isomers. 3-Alkyl-3-phenylcyclopropenes 1i–k could also be employed in the present ring-opening gem-silylborylation reaction to give products 3i–k in moderate yields of 30–48%, and the selectivity toward E isomers was found to be higher with smaller alkyl groups (up to E/Z = 84/16). In contrast, 3,3-dialkylcyclopropene 1l was found to give 1,2-silylborylation product 5la without ring-opening. As shown for the reaction using 2.0 mmol of 1g or 1j as the substrate, the present reactions were readily conducted on a preparative scale as well. With regard to the silylboron reagents, trialkylsilylboronates such as 2b and 2c were also applicable for the reaction of cyclopropene 1a to give corresponding (1-silyl)allylboronates 3ab and 3ac in high yields by raising the reaction temperature to 60 °C (eqn (1)).
:1 mixture of E/Z isomers. 3-Alkyl-3-phenylcyclopropenes 1i–k could also be employed in the present ring-opening gem-silylborylation reaction to give products 3i–k in moderate yields of 30–48%, and the selectivity toward E isomers was found to be higher with smaller alkyl groups (up to E/Z = 84/16). In contrast, 3,3-dialkylcyclopropene 1l was found to give 1,2-silylborylation product 5la without ring-opening. As shown for the reaction using 2.0 mmol of 1g or 1j as the substrate, the present reactions were readily conducted on a preparative scale as well. With regard to the silylboron reagents, trialkylsilylboronates such as 2b and 2c were also applicable for the reaction of cyclopropene 1a to give corresponding (1-silyl)allylboronates 3ab and 3ac in high yields by raising the reaction temperature to 60 °C (eqn (1)).

As previously mentioned, (1-silyl)allylboronates are known to be transformed into various products with high selectivity.3d As a brief demonstration, we examined the selective conversion of either boron or silicon moiety of compounds 3. For example, oxidation of the carbon–boron bond of 3ga was achieved by the reaction with sodium peroxoborate to give allylic alcohol 6 in 68% yield with retaining the silyl group (Scheme 2a).5c,12 A one-pot sequential process was also possible to prepare compound 6 from cyclopropene 1g in a comparable overall yield (Scheme 2b). On the other hand, allylic desilylative protonation of 3ja selectively took place under acidic conditions to give alkenylboronate 7 in 67% yield (Scheme 2c).5c
 | (1) |
 | ||
| Scheme 2 Transformations of (1-silyl)allylboronates 3. | ||
A proposed catalytic cycle for the reaction of cyclopropene 1a with silylboronate 2a is illustrated in Scheme 3. Initially, coordination of KOtBu to silylboronate 2a gives borate intermediate X,7b which is presumably in equilibrium with dissociated dimethyl(phenyl)silylpotassium and tBuOB(pin).7 Then, the silicon nucleophile attacks the alkene moiety of cyclopropene 1a, resulting in the formation of silylated cyclopropylpotassium intermediate Y. Subsequent ring-opening of this intermediate takes place with the release of the ring-strain to give π-allylpotassium Z. Finally, borylation with tBuOB(pin) proceeds at the silicon-substituted allylic carbon of Z, leading to the formation of (1-silyl)allylboronate 3aa along with regeneration of KOtBu.13
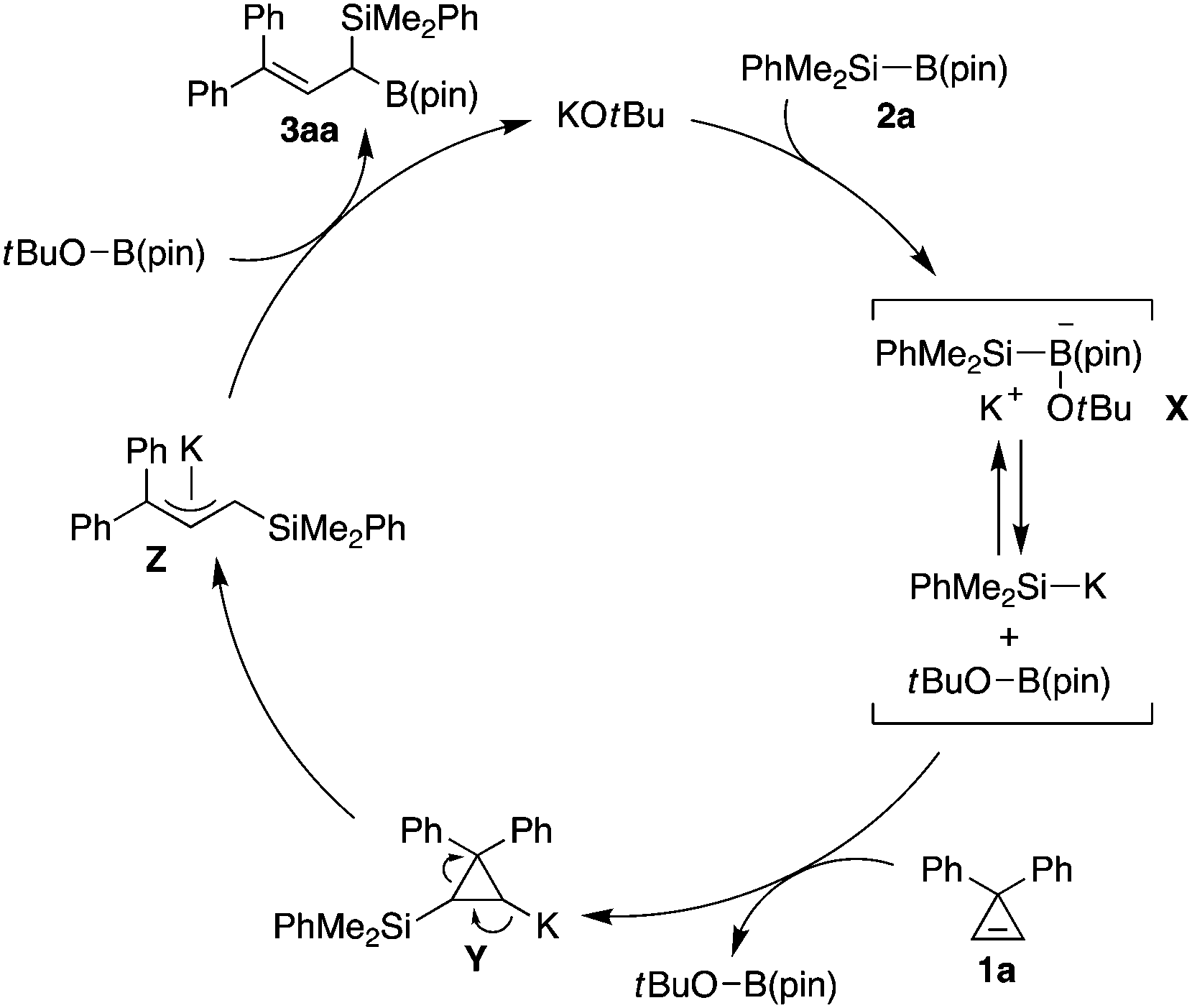 | ||
| Scheme 3 Proposed catalytic cycle for the synthesis of 3aa from 1a and 2a. | ||
To probe the feasibility of the above-mentioned catalytic cycle, a detailed reaction pathway was investigated by the DFT calculations for the reaction of 1a with Me3SiB(pin) in the presence of KOtBu at the M06/6-31+G(d) level of theory with solvation effect of THF (SMD) (see the ESI† for details). We assessed the Gibbs energy changes at standard conditions (298.15 K and 1 atm), and the resulting Gibbs energy profiles are depicted in Scheme 4.
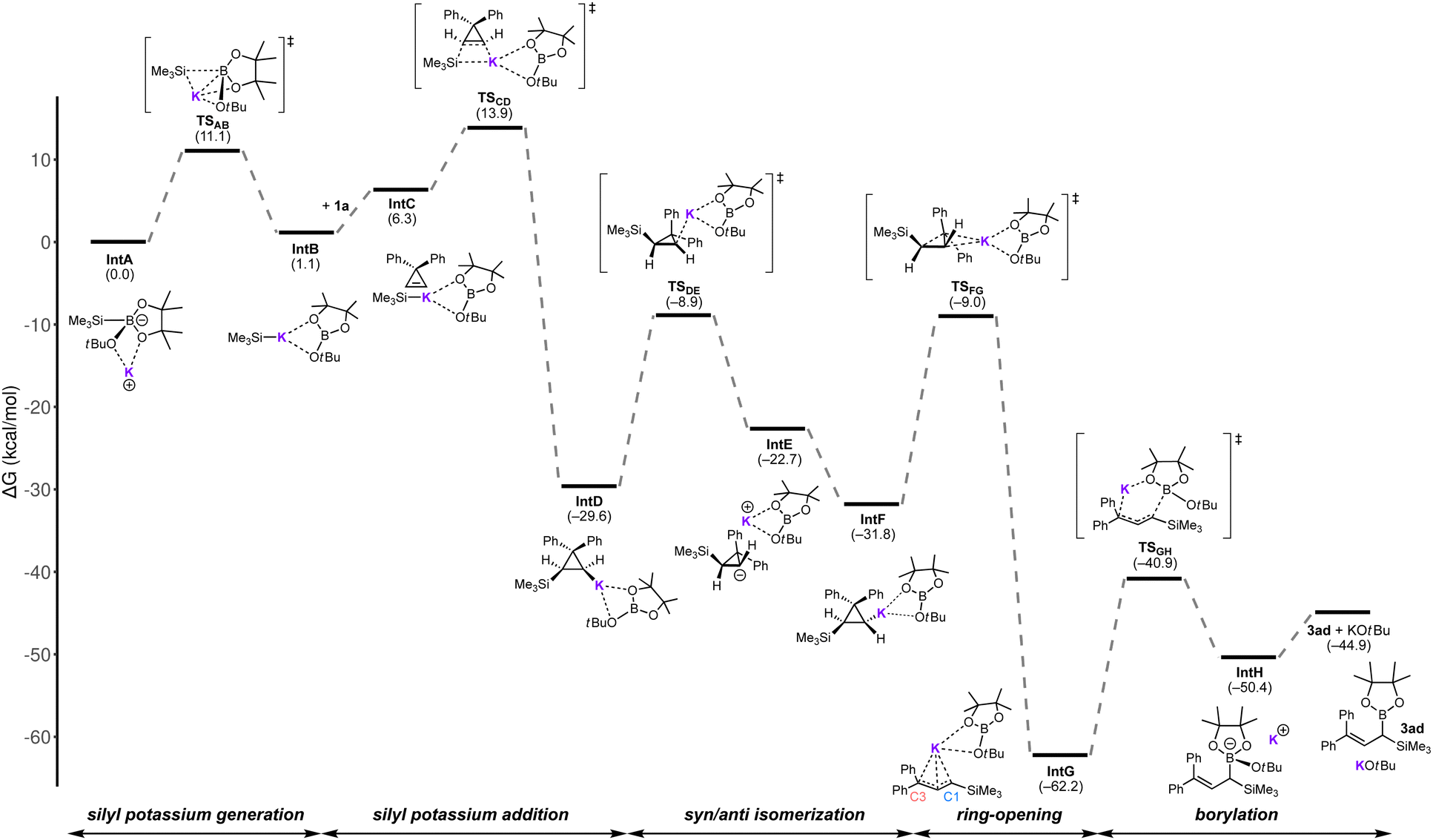 | ||
| Scheme 4 Calculated energy diagram for the KOtBu-catalyzed gem-silylborylation of 1a with trimethylsilylboronic acid pinacol ester. | ||
At first, borate intermediate IntA formed by the coordination of KOtBu to trimethylsilylboronate is converted to silylpotassium IntB with a Gibbs free energy change  of 1.1 kcal mol−1 through TSAB with activation energy
of 1.1 kcal mol−1 through TSAB with activation energy  of 11.1 kcal mol−1.7 Subsequently, the alkene of 1a coordinates to IntB to give IntC, which undergoes addition of the silyl anion to form cyclopropylpotassium species IntD through four-membered syn-addition transition state TSCD
of 11.1 kcal mol−1.7 Subsequently, the alkene of 1a coordinates to IntB to give IntC, which undergoes addition of the silyl anion to form cyclopropylpotassium species IntD through four-membered syn-addition transition state TSCD . This exergonic step is likely driven by the substantial release of the strain energy of a cyclopropene ring (55.2 kcal mol−1 for cyclopropene vs. 27.5 kcal mol−1 for cyclopropane).14
. This exergonic step is likely driven by the substantial release of the strain energy of a cyclopropene ring (55.2 kcal mol−1 for cyclopropene vs. 27.5 kcal mol−1 for cyclopropane).14
The ring-opening of syn-isomer IntD was found to be energetically unfavorable ( ; see Fig. S2 in the ESI†), and the syn/anti-isomerization of IntD through TSDE yields anti-isomer IntE
; see Fig. S2 in the ESI†), and the syn/anti-isomerization of IntD through TSDE yields anti-isomer IntE , which then relaxes to IntF
, which then relaxes to IntF . IntF undergoes the ring-opening to give π-allylpotassium IntGvia rate-determining TSFG
. IntF undergoes the ring-opening to give π-allylpotassium IntGvia rate-determining TSFG , following the Woodward–Hoffmann rules in a conrotatory electrocyclic reaction of a 4-electron system.15 Ring-opened IntG is calculated to be thermodynamically stable presumably due to the α-effect by the silyl group16 and the resonance effect by the two aryl substituents. Allylpotassium species IntG then attacks the neighboring borate to form gem-silylborylated IntHvia six-membered transition state TSGH
, following the Woodward–Hoffmann rules in a conrotatory electrocyclic reaction of a 4-electron system.15 Ring-opened IntG is calculated to be thermodynamically stable presumably due to the α-effect by the silyl group16 and the resonance effect by the two aryl substituents. Allylpotassium species IntG then attacks the neighboring borate to form gem-silylborylated IntHvia six-membered transition state TSGH , which then regenerates the KOtBu catalyst through the liberation of product 3ad for the next cycle. The natural population analysis (NPA) of IntG revealed that the C1-position, bearing a SiMe3 group, exhibits a higher negative charge compared to the C3-position (C1: −0.973 vs. C3: −0.324). This observation implies that the carbon–boron formation preferentially takes place at the C1-position.
, which then regenerates the KOtBu catalyst through the liberation of product 3ad for the next cycle. The natural population analysis (NPA) of IntG revealed that the C1-position, bearing a SiMe3 group, exhibits a higher negative charge compared to the C3-position (C1: −0.973 vs. C3: −0.324). This observation implies that the carbon–boron formation preferentially takes place at the C1-position.
In summary, we developed a KOtBu-catalyzed ring-opening gem-silylborylation of cyclopropenes with silylboronates to give (1-silyl)allylboronates under simple and mild conditions. The utility of the resulting products was briefly examined, and the reaction mechanism was probed in detail using DFT calculations. Future studies will be directed toward further development of silylborylation using silylboronates under base catalysis.
Support has been provided in part by Toray Science Foundation (R. S.), JSPS KAKENHI Grant Number JP24H01856 (Grant-in-Aid for Transformative Research Areas (A) Green Catalysis Science; R. S.), and JST, the establishment of university fellowships towards the creation of science technology innovation, Grant Number JPMJFS2125 (H. M.). We thank Mr Daigo Hayashi at Osaka University for the measurement of high-resolution mass spectra. The computations were performed using the Research Center for Computational Science, Okazaki, Japan (Projects: 23-IMS-C165, 24-IMS-C167).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews: (a) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed; (b) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed; (c) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS; (d) R. W. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 555 CrossRef; (e) C. Diner and K. J. Szabó, J. Am. Chem. Soc., 2017, 139, 2 CrossRef CAS PubMed; (f) H.-X. Huo, J. R. Duvall, M.-Y. Huang and R. Hong, Org. Chem. Front., 2014, 1, 303 RSC; (g) J. W. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732 CrossRef CAS PubMed; (h) A. Hosomi, Acc. Chem. Res., 1988, 21, 200 CrossRef CAS; (i) I. Fleming, Chem. Soc. Rev., 1981, 10, 83 RSC.
- For selected recent examples: (a) M. Z. Liang and S. J. Meek, J. Am. Chem. Soc., 2020, 142, 9925 CrossRef CAS PubMed; (b) M. Berger, D. Carboni and P. Melchiorre, Angew. Chem., Int. Ed., 2021, 60, 26373 CrossRef CAS PubMed; (c) Z.-N. Yang, H. Rao, Y. Yin, S. Mu, Z. Jia and H. Ding, Org. Lett., 2024, 26, 3524 CrossRef CAS PubMed; (d) M. Ghosh, S. Sahu, S. Saha and M. S. Maji, Chem. Sci., 2024, 15, 1789 RSC.
- (a) D. J. S. Tsai and D. S. Matteson, Organometallics, 1983, 2, 236 CrossRef CAS; (b) M. Shimizu, H. Kitagawa, T. Kurahashi and T. Hiyama, Angew. Chem., Int. Ed., 2001, 40, 4283 CrossRef CAS; (c) J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 9893 CrossRef CAS PubMed; (d) J. Park, Y. Jung, J. Kim, E. Lee, S. Y. Lee and S. H. Cho, Adv. Synth. Catal., 2021, 363, 2371 CrossRef CAS.
- For selected recent examples: (a) R. Tong, S. Liu, C. Zhao, D. Jiang, L. Gao, W. Wang, B. Ye and Z. Song, Org. Lett., 2022, 24, 7822 CrossRef CAS PubMed; (b) Y. Jung and S. H. Cho, Synlett, 2023, 2165 CAS; (c) Y. Jung, S. Y. Yoo, Y. Jin, J. You, S. Han, J. Yu, Y. Park and S. H. Cho, Angew. Chem., Int. Ed., 2023, 62, e202218794 CrossRef CAS PubMed.
- (a) H. Yatagai, Y. Yamamoto and K. Maruyama, J. Am. Chem. Soc., 1980, 102, 4548 CrossRef CAS; (b) M. Chen and W. R. Roush, Org. Lett., 2013, 15, 1662 CrossRef CAS PubMed; (c) M. Huang, Y. Zhao, C. Zhang and S. F. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202203343 CrossRef CAS PubMed; (d) C. Bomio, M. Kabeshov, A. Lit, S. Lau, J. Ehlert, C. Battilocchio and S. Ley, Chem. Sci., 2017, 8, 6071 RSC; (e) C. Wu, Z. Bao, X. Xu and J. Wang, Org. Biomol. Chem., 2019, 17, 5714 RSC; (f) J. Kim, E. Lee and S. H. Cho, Asian J. Org. Chem., 2019, 8, 1664 CrossRef CAS; (g) M. Murakami, I. Usui, M. Hasegawa and T. Matsuda, J. Am. Chem. Soc., 2005, 127, 1366 CrossRef CAS PubMed.
- For selected recent examples using silylboronates as the nucleophilic reagent: (a) E. Jeong, J. Heo, S. Jin, D. Kim and S. Chang, ACS Catal., 2022, 12, 4898 CrossRef CAS; (b) H. Zhang, L. Jiang, M. Yang and Y. Liu, Org. Chem. Front., 2023, 10, 3598 RSC; (c) L. Gao, X. Liang, L. He, G. Li, S. Chen, J. Cao, J. Ma, G. Wang and S. Li, Chem. Sci., 2023, 14, 11881 RSC.
- (a) A. Kawachi, T. Minamimoto and K. Tamao, Chem. Lett., 2001, 1216 CrossRef CAS; (b) H. Ito, Y. Horita and E. Yamamoto, Chem. Commun., 2012, 48, 8006 RSC; (c) R. Shintani, R. Fujie, M. Takeda and K. Nozaki, Angew. Chem., Int. Ed., 2014, 53, 6546 CrossRef CAS PubMed; (d) K. Kojima, Y. Nagashima, C. Wang and M. Uchiyama, ChemPlusChem, 2019, 84, 277 CrossRef CAS PubMed; (e) X.-W. Liu, C. Zarate and R. Martin, Angew. Chem., Int. Ed., 2019, 58, 2064 CrossRef CAS PubMed.
- (a) J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010 RSC; (b) J. R. Wilkinson, C. E. Nuyen, T. S. Carpenter, S. R. Harruff and R. Van Hoveln, ACS Catal., 2019, 9, 8961 CrossRef CAS.
- S. Kishida, M. Takano, T. Sekiya, Y. Ukaji and K. Endo, J. Org. Chem., 2022, 87, 14833 CrossRef CAS PubMed.
- Some of these compounds are somewhat unstable for chromatographic purification, resulting in lower isolated yields compared with the NMR yields.
- CCDC 2340586 contains the supplementary crystallographic data for this paper†.
- H. C. Brown and G. A. Zweifel, J. Am. Chem. Soc., 1959, 81, 247 CrossRef CAS.
- The radical mechanism is less likely based on the amount of KOtBu employed and the trace oxygen test: (a) B. Cui, Y. Tian, Y. Gao, S. Hua, Y. Shi and C. Cao, J. Org. Chem., 2023, 88, 11173 CrossRef CAS PubMed; (b) M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 1970 CrossRef CAS PubMed; (c) J. Zhou, B. Jiang, Y. Fujihira, Z. Zhao, T. Imai and N. Shibata, Nat. Commun., 2021, 12, 3749 CrossRef CAS PubMed.
- (a) P. V. R. Schleyer, J. E. Williams and K. R. Blanchard, J. Am. Chem. Soc., 1970, 92, 2377 CrossRef CAS; (b) R. Vicente, Chem. Rev., 2021, 121, 162 CrossRef CAS PubMed.
- (a) R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395 CrossRef CAS; (b) R. Huisgen and P. Eberhard, J. Am. Chem. Soc., 1972, 94, 1346 CrossRef CAS; (c) P. K. Chou, G. D. Dahlke and S. R. Kass, J. Am. Chem. Soc., 1993, 115, 315 CrossRef CAS.
- (a) D. M. Wetzel and J. I. Brauman, J. Am. Chem. Soc., 1988, 110, 8333 CrossRef CAS; (b) E. A. Brinkman, S. Berger and J. I. Brauman, J. Am. Chem. Soc., 1994, 116, 8304 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2340586. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01336k |
| This journal is © The Royal Society of Chemistry 2024 |