
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chiral trimethyl lock based on the vicinal disubstituent effect: prolonged release of camptothecin into cancer cells†
Silvia
Venturi
a,
Ferdinando
Chiaradonna
 b,
Francesco G.
Gatti
*a,
Barbara
La Ferla
*c,
Roberta
Palorini
b and
Barbara
Zerbato
b
b,
Francesco G.
Gatti
*a,
Barbara
La Ferla
*c,
Roberta
Palorini
b and
Barbara
Zerbato
b
aDepartment Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico of Milan, Piazza Leonardo da Vinci 32, 20133 Milano, Italy. E-mail: francesco.gatti@polimi.it
bDepartment of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milano, Italy
cDepartment of Earth and Environmental Science, University of Milano-Bicocca, Piazza della Scienza 1, 20126 Milano, Italy. E-mail: barbara.laferla@unimib.it
First published on 27th May 2024
Abstract
Synthesis and in vitro testing of a prodrug designed for the controlled delivery of the anticancer drug camptothecin within pancreatic cancer cells are reported. Our study reveals a non-conventional pharmacokinetic release characterized by an exponential pattern before reaching the half-life (t1/2) and a linear pattern thereafter. The release mechanism was triggered either by hydrolytic enzymes and/or by the acid microenvironment of cancer cells.
In the early 1970s, Cohen et al. investigated the acid-catalysed lactonization of a series of methyl substituted o-hydroxyphenyl propionic acids, intending to find a model reaction having rate constants comparable to those of enzymatic transformations.1 They found that the reaction rate is strongly dependent on the number of steric repulsions between the methyl substituents of the linear precursors (Fig. 1A). In particular, it was shown that the gem-dimethyl acid ring closes much more quickly than its unsubstituted homologue (Thorpe–Ingold effect2) and that the buttressing effect3 exerted by the C(3) aromatic methyl group on the two geminal methyl substituents results in an astonishing acceleration of the reaction rate.4
 | ||
| Fig. 1 (A) Ring-closure of methyl substituted hydrocoumarin derivatives. (B) Drug release mechanism of TML-based prodrugs. (C) Thorpe–Ingold effect and vic-disubstituent effect in the lactonization of γ and δ-hydroxyesters. | ||
Over the years, many other systems like Cohen's archetype (commonly referred to as “trimethyl lock”, TML) have been designed, finding important applications in the field of medicinal chemistry (Fig. 1B).5 Indeed, it has been shown that prodrugs based on TMLs allow a quasi-instantaneous release of biologically active compounds upon specific chemical, photochemical or enzymatic triggers. For such systems, the release process terminates after a period equal to 4–5 times the half-life (t1/2); however, there are several drugs whose local dosage concentration is crucial for therapeutic efficacy, and for which quantitative releases performed in a too short time might be detrimental. Conversely, systems in which it is possible to modulate the release rate should increase their therapeutic window (dosage range, in which the drug operates effectively without having toxic effects).6 So far, prolonged releases have been achieved only by encapsulating the drug in a nanogel or polymeric matrix.7
In this regard, we envisaged that a chiral TML should be able to fulfil such a task, since the release would not be longer controlled by a single rate constant, as in Cohen's system, but by the combination of more rate constants, all depending on the relative configuration of each diastereoisomer.
Recently, we studied the acid-catalysed cyclization of a series of alkyl substituted γ- and δ-hydroxy esters and we found that the cyclization rate depends on the relative stereochemistry of the vic-disubstituted precursors (vic-disubstituent effect), regardless of the catalyst, the leaving group and the solvent used.8 More precisely, in the case of γ-hydroxy esters, the syn dimethyl substituted diastereoisomer showed a kinetic behaviour quite similar to its gem-dimethyl constitutional isomer, whereas, the anti diastereoisomer ring-closed to the trans lactone almost 20 times faster; analogously, even for the δ-hydroxy esters a vic-disubstituent effect was observed (Fig. 1C).
Thus, keeping this in mind, we studied the trifluoro acetic acid (TFA) catalysed ring-closure of the trimethyl substituted δ-hydroxy i-propyl ester 1 (chiral-TML) to lactone 2 (Fig. 2A). Ester 1 has three contiguous stereogenic centres and it exists as a mixture of four diastereoisomers (syn,syn-1, syn,anti-1, anti,syn-1 and anti,anti-1); the synthesis of 1 was accomplished in seven steps from ethyl crotonate and 2,4-pentadione and in an overall yield of 21% (for more details see ESI†).
 | ||
| Fig. 2 (A) TFA-catalysed ring-closure of the diastereomeric mixture of the trimethyl substituted δ-hydroxy ester 1 in CDCl3 at 302 K. The relative stereochemical configurations of lactones were assigned by 1H-NMR in C6D6. (B) 1H-NMR (400 MHz) spectra of the CHOH signal at different times: (a) [TFA] = 4.4 10−5 M; (b) [TFA] = 1.70 10−4 M. | ||
The TFA-catalysed lactonization of 1 was monitored by 1H-NMR spectroscopy (CDCl3, 302 K), integrating the CHOH proton signal of each diastereoisomer (Fig. 2B). The relative stereochemistry of each stereoisomer of 1 was indirectly assigned from the configuration of the corresponding lactone diastereoisomer, which in turn was assigned by measuring the JH(6)–H(5) coupling constant of the H(6) signal in C6D6 (see ESI†).
The analysis of the data pointed to a pseudo first-order rate law in the hydroxy ester concentration [E]. The reaction rate constant kobs (s−1) was obtained by linear regression of ln = [E]/[E]0vs. time, and the second order rate constant kr (s−1 M−1) was calculated from the relation kobs = kr[TFA] (for the kinetic progress curves and the linear regression plots see the ESI†).
In Fig. 2A are reported the reaction rate constants (kr) and the free activation energy barriers (δΔG‡) relative to the lactonization of the slowest isomer, i.e. the anti,syn-1. The kinetic study shows that the isomer anti,syn-1 ring-closes to cis,cis-2 (lactone with the highest ring strain energy) roughly sixty times slower than the fastest isomer (syn,anti-1), while the reaction rate of syn,syn-1 does not change much (kr = 1.5). A much larger difference in reactivity was observed with the anti,anti-1 isomer, which cyclizes about eight times faster than syn,syn-1.
Overall, these results show that the vic-effect exerted by the coupled C(4)Me and C(5)Me methyl groups is larger than that generated by the C(3)Me and C(4)Me substituents. But more interestingly, the sum of these two contiguous vic-effects enables the release of the i-propanol leaving group with a unique kinetic profile. Fig. 3 shows the i-PrOH progressive curve of each diastereoisomer of 1 (dashed lines) compared to that of the equimolar diastereomeric mixture of 1 (solid line).
 | ||
| Fig. 3 Kinetic progress curves of i-PrOH release relative to the TFA catalysed ring-closure of 1 in CDCl3; dashed line for each diastereoisomer, and solid line for the equimolar diastereomeric mixture of 1. | ||
The anti,syn isomer releases i-PrOH very rapidly (t1/2 ≈ 230 s), and its kinetic behaviour is comparable to that of the typical TMLs (very fast release). Remarkably, the t1/2 of the diastereomeric mixture has the same order of magnitude as that of the anti,anti isomer (2300 s vs. 1250 s), but after t1/2 the formation rates of i-PrOH are very different. In this phase, the formation of i-PrOH from the fastest isomers is virtually completed, and the kinetics is controlled mainly by the rate constants of the slowest isomers (i.e. anti,syn and syn,syn), affording a prolonged release of i-PrOH (slow release, which is still persistent even after 8 × t1/2). In summary, the kinetic profile of the TFA-catalysed lactonisation of an equimolar diastereomeric mixture of 1 is characterised by two well-distinct periods: before t1/2, in which we observe an exponential increase of i-PrOH (fast release), and after t1/2, when the release rate decreases more gradually (prolonged release with a linear pattern).
Now, according to our ongoing research program on anticancer therapy with novel drugs,9 we decided to exploit the chiral-TML for the release of camptothecin10 (CPTOH, Scheme 1). Indeed, although the CPTOH has proven to be an efficient anticancer agent, a few ways to mitigate its severe side effects against healthy cells in chemotherapeutic treatments have been developed.
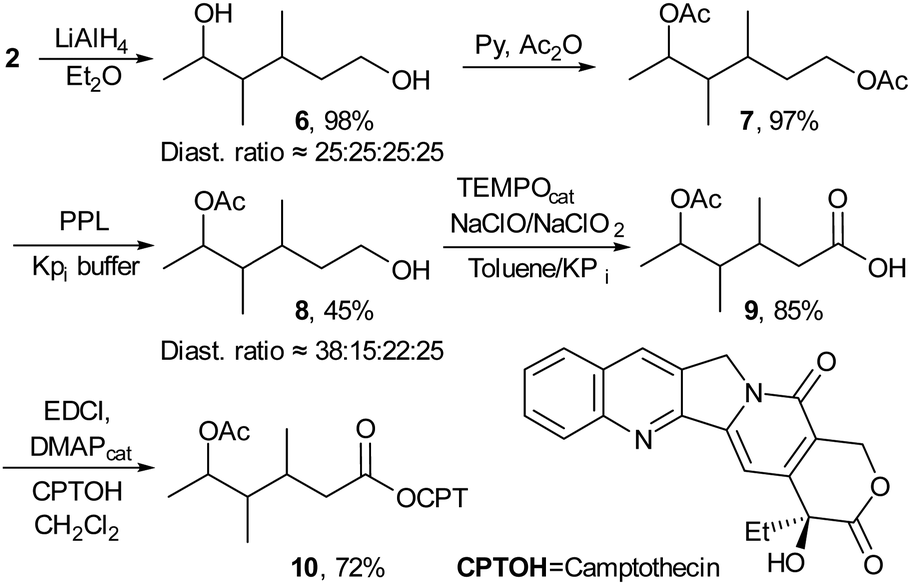 | ||
| Scheme 1 Synthesis of the chiral-TML based camptothecin prodrug. | ||
For example, CPTOH was coupled with a benzoquinone derivative through its tertiary alcohol and the release was triggered by a DT-diaphorases enzyme, which reduced quinone into the reactive hydroquinone intermediate.11 Alternatively, since the tumour tissues are known to be slightly acidic (pH ≈ 6–7),12 pH-responsive prodrugs have been developed as well.13 In this context, we designed the chiral-TML-based prodrug 10, with the drug release triggered by a chemo and/or enzymatic hydrolysis (Fig. 4A) of the acetyl group.
 | ||
| Fig. 4 (A) Acid/enzymatic triggered release mechanism of CPTOH from 10. (B) HPLC of acid triggered hydrolysis of 10 (96 h); (C) ESI-MS spectra of Int, collected from elution. | ||
The synthesis of 10 was accomplished in five steps and an overall yield of 26% (Scheme 1). First, the lactone 2 was reduced with LiAlH4 affording diol 6 (98% yield), which was acetylated to the diester 7 with Ac2O in a quantitative yield. Hydrolysis of 7, catalysed by porcine pancreatic lipase (PPL) gave mainly the primary alcohol 8,14 which was separated from the secondary alcohol by column chromatography, but a slightly different diastereomeric ratio compared to the starting diol was observed (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:25:25 vs. 38:15:22:25, by 13C-NMR). Then, the primary alcohol was oxidised to the carboxylic acid 9 in 85% yield, using TEMPO and NaClO/NaClO2.15 In the last step, 10 was obtained by coupling of acid 9 with the tertiary alcohol of CPTOH, using the EDCI condensing agent, in a yield of 72% after purification by column chromatography.
:25:25:25 vs. 38:15:22:25, by 13C-NMR). Then, the primary alcohol was oxidised to the carboxylic acid 9 in 85% yield, using TEMPO and NaClO/NaClO2.15 In the last step, 10 was obtained by coupling of acid 9 with the tertiary alcohol of CPTOH, using the EDCI condensing agent, in a yield of 72% after purification by column chromatography.
Next, we focused our attention on setting up the trigger through enzymatic hydrolysis of the acetyl group to form the reactive intermediate Int (Fig. 4A). In this regard, a set of commercially available lipases and esterases were tested in a buffer solution (KPi), disappointingly, under these conditions, the prodrug remained highly stable (see ESI†). However, these results do not preclude the possibility of other enzymes capable of hydrolyzing the acetyl group of 10, vide infra. As an alternative approach, the prodrug was submitted to chemical hydrolysis (buffer at pH = 5, 35 °C), at the pH that can be found in the lysosomes,16 and the reaction-time course was monitored by HPLC and TLC (see ESI†). After 20 hours we detected the release of CPTOH (tR 7.3 min) along with the appearance of two new chromatographic peaks (tR 12.0 min and 12.5 min, Fig. 4B) corresponding to the diastereoisomers of Int (Fig. 4B), as confirmed by ESI-MS analysis (Fig. 4C). Even in this case, the reaction time-course initially revealed a rapid release of CPTOH, which slowed down after 100 hours (see ESI†).
Finally, in vitro tests were carried out to assess the prodrug effects on the viability of pancreatic cancer cells. MIAPaCa-2 cell cultures treated with equimolar concentrations (200 nM) of CPTOH, 10 and lactone 2, were compared at different time points (24, 48, 72 and 96 h) with untreated cells; the results after 96 h are shown in Fig. 5 and those of previous time-points in the ESI† (Fig. S11). The treatment with 10, as compared to the untreated cells, induced a substantial reduction of the cell number at all the time points analysed (by Trypan blue assay, upper histogram of Fig. 5A and Fig. S11, ESI†), concomitant to an increased cell death (see lower histogram and Fig. S11, ESI†), indicating the ability of this prodrug to deliver CPTOH in pancreatic cancer cells. Indeed, according to the literature, the esterification of the tertiary alcohol of CPTOH dramatically affects its biological activity.10a To further confirm the effective release of CPTOH into the cellular microenvironment, the extraction and the mass spectroscopy multiple reaction monitoring analysis (MS-MRM) of cellular metabolites was carried out on MIAPaCa-2 cell cultures treated with 10 (200 nM) after 48 h. Both 10 and CPTOH were detected, suggesting the permeation of the prodrug through the cellular membrane and the subsequent release of the anticancer drug (Fig. 6). Notably, the MRM chromatogram of 10 from the cellular extract (Fig. 6B), clearly shows a net change in the diastereoisomeric distribution with respect to the chromatogram of standard 10 (Fig. 6A), suggesting that in the pancreatic cells a stereoselective enzymatic hydrolysis of 10 is occurring.
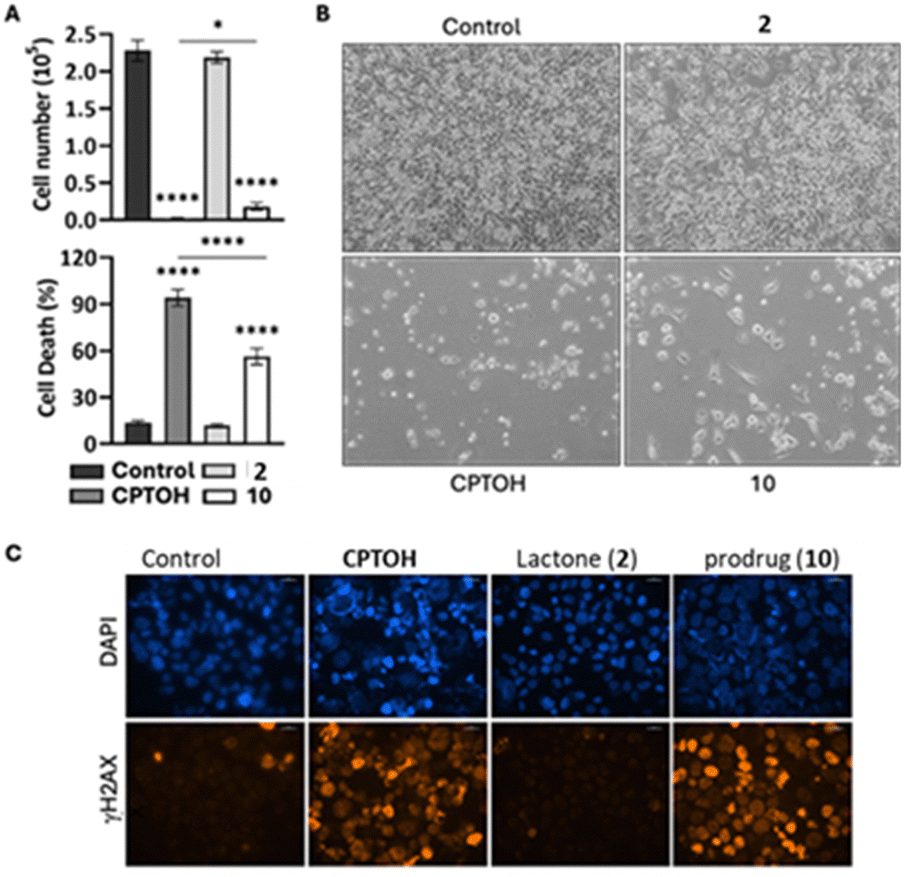 | ||
| Fig. 5 MIAPaCa-2 cells treated for 96 hours with CPTOH, 2, or 10 (200 nM). (A) Cell number and cell death (% of total population). (B) Microscope images (4×). (C) Immunofluorescence using fluorescence microscopy images (40×): DAPI (nuclei) and γH2AX (DNA damage). | ||
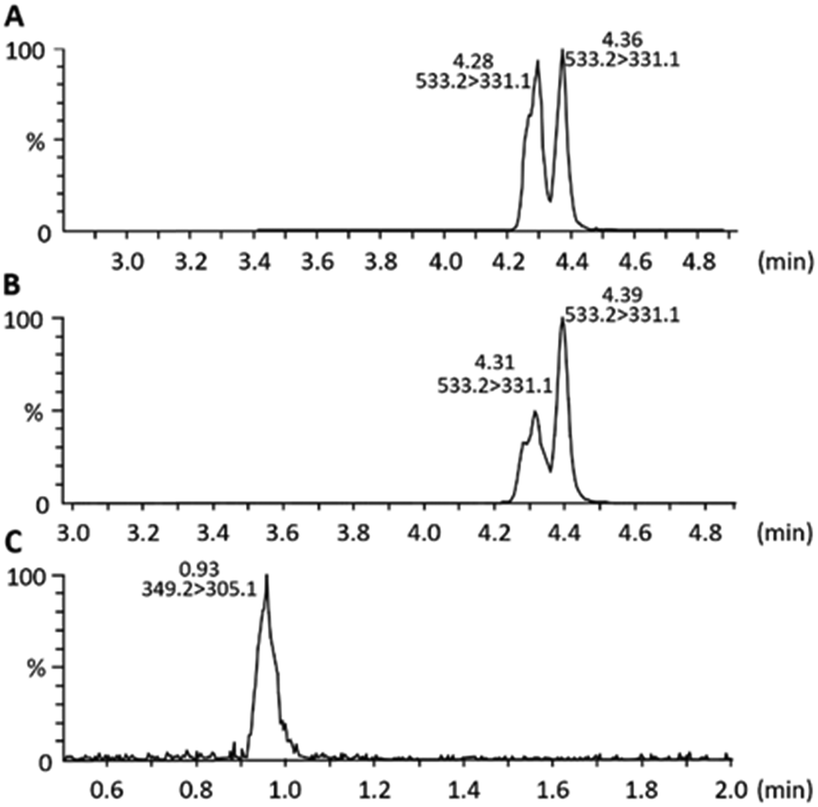 | ||
| Fig. 6 MS-MRM analysis: transitions monitored for 10m/z 533.2 → m/z 333.1, (A) reference compound, and (B) cellular extract; transitions monitored for CPTOHm/z 349.1 → m/z 305.1 cellular extract (C). | ||
The phosphorylation of nuclear histone H2AX (γH2AX) is an important indicator of DNA damage caused by chemotherapeutics, such as CPTOH.17 As it can be seen from the images of fluorescence microscopy, 10 was able to stimulate the expression of γH2AX as well as CPTOH, further confirming that it retains the same action mechanism (Fig. 5C). In addition, lactone 2 showed no toxicity, as evidenced by the cell viability test and the γH2AX level (Fig. 5A and C and ESI,† Fig. S11), comparable to the control. Indeed, its effect, in terms of cell number and cell death, was comparable to that observed in the untreated cells.
Morphological analysis under phase contrast microscopy further confirmed these results. The CPTOH treatment, starting from 48 h, drastically reduced the cell number and significantly changed the cell morphology inducing the appearance of a large proportion of detached cells with a clear loss of the cell membrane structure, showing massive cell death (Fig. 5B). Such behaviour was less pronounced in the cells treated with prodrug 10. We reckon that this difference might be ascribed to the time-prolonged release of camptothecin, rather than to the reduced anticancer activity of 10 itself since its tertiary alcohol is esterified, unlike all structurally modified active analogues of CPTOH.10a
In conclusion, the unique kinetic behaviour of a chiral TML is exploitable for the design of new prodrugs, such as 10. The delivery of the biologically active molecules can be triggered by the acid environment of cancer cells or by enzymatic hydrolytic activities, which are still to be identified. However, the release initially occurs very rapidly (acute treatment) and after t1/2 gradually slowdowns ensuring a prolonged level of drug concentration (maintenance of drug dose level), as a strategy to mitigate its severe side effects against healthy cells.
Conceptualization and methodology FGG, BLF, and FC; investigation, SV, RP, BZ; writing FGG, BLF, FC and SV; project administration, FGG, BLF.
SV-FGG and FC-BLF thank POR-Lombardia, VIPCAT project (ID 228775) and Fondo di Ateneo-Quota Competitiva (#ATEQC-0006 and #ATEQC-0001), respectively, for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Milstien and L. A. Cohen, Proc. Natl. Acad. Sci. U. S. A., 1970, 67, 1143–1147 CrossRef CAS PubMed.
- M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef CAS PubMed.
- P. G. Sammes and D. J. Weller, Synthesis, 1995, 1205–1222 CrossRef CAS.
- The early reported rate constants have been later revised: M. Caswell and G. L. Schmir, J. Am. Chem. Soc., 1980, 102, 4815–4821 CrossRef CAS.
- (a) M. L. Levine and R. T. Raines, Chem. Sci., 2012, 3, 2412 RSC; (b) O. A. Okoh and P. Klahn, ChemBioChem, 2018, 19, 1668 CrossRef CAS PubMed.
- L. Brunton and K. L. Parker, Goodman&Gilman's Manual of Pharmacology and Therapeutics, MacGraw Hill, 2007.
- (a) F. Seidi, R. Jenjob and D. Crespy, Chem. Rev., 2018, 118, 3965 CrossRef CAS PubMed; (b) V. X. Truong, F. Li, F. Ercole and J. S. Forsythe, Chem. Commun., 2017, 53, 12076 RSC.
- (a) E. Brenna, F. Distante, F. G. Gatti and G. Gatti, Catal. Sci. Technol., 2017, 7, 1497 RSC; (b) E. Brenna, F. Dalla Santa, F. G. Gatti, G. Gatti and D. Tessaro, Org. Biomol. Chem., 2019, 17, 813 RSC.
- F. Ricciardiello, Y. Gang, R. Palorini, Q. Li, M. Giampà, F. Zhao, L. You, B. La Ferla, H. De Vitto, W. Guan, J. Gu, T. Zhang, Y. Zhao and F. Chiaradonna, Oncogene, 2020, 39, 4103 CrossRef CAS PubMed.
- (a) C. J. Thomas, N. J. Rahier and S. M. Hecht, Bioorg. Med. Chem., 2004, 12, 1585 CrossRef CAS PubMed; (b) Y.-Q. Liu, W.-Q. Li, S. L. Morris-Natschke, K. Qian, L. Yang, G.-X. Zhu, X.-B. Wu, A.-L. Chen, S.-Y. Zhang, X. Nan and K.-H. Lee, Med. Res. Rev., 2015, 35, 735 Search PubMed.
- P. Liu, J. Xu, D. Yan, P. Zhang, F. Zeng, B. Li and S. Wu, Chem. Commun., 2015, 51, 9567 RSC.
- C. Corbet and O. Feron, Nat. Rev. Cancer, 2017, 17, 577 CrossRef CAS PubMed.
- S.-Y. Li, L. H.-Liu, H.-Z. Jia, W.-X. Qiu, L. Rong, H. Cheng and X.-Z. Zhang, Chem. Commun., 2014, 50, 11852 RSC.
- G. Pedrocchi-Fantoni and S. Servi, J. Chem. Soc., Perkin Trans. 1, 1992, 1029 RSC.
- M. Zhao, J. Li, E. Mano, Z. Song, D. M. Tschaen, E. J. J. Grabowski and P. J. Reider, J. Org. Chem., 1999, 64, 2564 CrossRef CAS.
- A. A. H. Ponsford, T. A. Ryan, A. Raimondi, E. Cocucci, S. A. Wycislo, F. Fröhlich, L. E. Swan and M. Stagi, Autophagy, 2021, 17, 1500 CrossRef CAS PubMed.
- T. Furuta, H. Takemura, Z.-Y. Liao, G. J. Aune, C. Redon, O. A. Sedelnikova, D. R. Pilch, E. P. Rogakou, A. Celeste, H. Tang Chen, A. Nussenzweig, M. I. Aladjem, W. M. Bonner and Y. Pommier, J. Biochem., 2003, 22, 20303 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization, copies of 1H-NMR and 13C-NMR spectra, progress curves and linear regressions of the kinetic study. See DOI: https://doi.org/10.1039/d4cc01220h |
| This journal is © The Royal Society of Chemistry 2024 |