
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical cyclopropanation in aqueous micellar media – experimental and theoretical studies†
Joseph P.
Milton
 a,
Adam
Milanowski
ab,
Martin
Andersson
*c and
Dorota
Gryko
*a
a,
Adam
Milanowski
ab,
Martin
Andersson
*c and
Dorota
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, Warsaw 01-224, Poland. E-mail: dorota.gryko@icho.edu.pl
bDepartment of Chemistry, Warsaw University of Technology, Noakowskiego 3, Warsaw 00-664, Poland
cCenter for Integrative Petroleum Research, King Fahd University of Petroleum and Minerals, Dhahran 31261, Kingdom of Saudi Arabia. E-mail: martin.andersson@kfupm.edu.sa
First published on 2nd April 2024
Abstract
While in nature, reactions occur in water-based confined compartments, for a long time, water has been often regarded as an unsuitable medium for organic reactions. We have, however, found that photochemical cyclopropanation of styrenes with diazo compounds or their precursors can be performed in micellar systems. COSMO-RS studies revealed that the reactivity correlates with the predicted critical micelle concentration (CMC), with higher CMC values delivering higher yields.
Organic solvents comprise the majority of waste in the chemical industry, which is believed to account for more than 60% by mass in the pharmaceutical sector.1 One way to minimise solvent waste is to implement water, which is non-toxic, non-flammable, cheap, safe,2 and enables a reaction to adhere more closely to the 12 principles of green chemistry.3 Of course, the prominent issue with water is the poor solubility of organic reagents in this medium, which leads to most chemists automatically ruling it out as a solvent choice;4 undoubtedly, there are examples where photochemical reactions in water can successfully take place though.5,6 To enhance the viability of reactions in water, a surfactant can be added, thus forming a micellar solution. They have been known for many years for their remarkable properties as solubilisers to dissolve compounds under aqueous conditions,1 enabling organic synthesis in water that may not be possible otherwise.7–14 With the reagents encapsulated inside of the micelle, the components of the reaction pre-organise themselves, which can also lead to increased selectivies.15 Moreover, the lifetimes of short-lived species, such as carbenes and radicals, are prolonged in micelles.9,16 Studies into the generation of UV-light in situ via triplet–triplet annihilation upconversion in micelles were recently reviewed by Næsborg and co-workers.17
Cyclopropanes are one of the top ten most frequently represented moieties in FDA-approved drugs, and hence greener approaches for their construction are of great benefit to the pharmaceutical sector.18,19 With the copious number of methods for their synthesis, diazo compounds are one of the most atom economical starting materials that can be used, since only the concomitant loss of dinitrogen is required to generate the reactive species under thermal, photochemical, or metal-catalysed conditions.20 Photochemical activation of diazo compounds often performs optimally with DCM, although it is notoriously toxic21 and poses a high risk to the environment.22 Using water would alleviate the toxicity issue and make these reactions safer, but reactions with carbenes in water pose an additional challenge as O–H insertion is always a viable side reaction.23
In turn, there are only a limited number of methods involving diazo compounds in water and most of them are metal-catalysed. Álvarez et al. used a copper catalyst to allow cyclopropanation and C–H insertion of diazo compounds in water,24 while the use of a water-soluble Ru-porphyrin enabled N–H insertion reactions in a buffer solution.25 Rh-catalysed lactam and lactone synthesis was also accomplished in this medium.26–28 Li's group showed that [2+1]-cycloadditions can be performed in water without any external stimuli with the diazo moiety simply cleaved as a leaving group.29
Only two reports describe photochemical reactions of diazo compounds in water. UV irradiation of various α-diazo acetamides leads to intramolecular C–H insertions to form an assortment of lactams.30 O–H insertion was also observed and the propensity towards hydroxylation was linked to the hydrophilicity of the diazo compound. Hydrophobic dibutyl acetamides formed lactams as the exclusive product, whereas acetamides bearing hydrophilic 2-methoxyethyl substituents reacted only with water. Later, Yan et al. showed the in situ formation of diazo compounds from hydrazones in the presence of triethylamine in water and their subsequent reaction with acetylenes, olefins, anilines, and thiols under blue LED irradiation.31
We hypothesised that photochemical cyclopropanation of diazo compounds could be performed in a micellar solution; in particular, Maaskant et al. has shown the beneficial effects of micelles for diazo compounds in an iron-catalysed process. In the presence of a cationic Fe-porphyrin in water, only traces of the desired cyclopropane were detected, whereas the addition of sodium dodecyl sulfate (SDS) increased the yields up to >99% in the best case.32
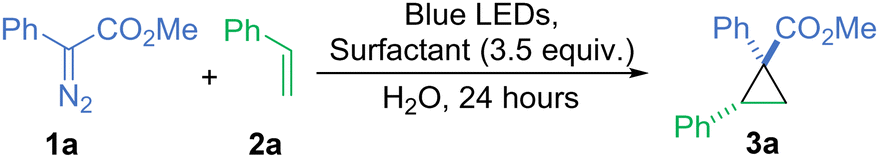
To begin our investigation, we tested the feasibility of methyl phenyldiazoacetate (1a) with styrene (2a) to form cyclopropane 3a in a variety of micellar solutions, of which, a solution of cationic dodecyl trimethylammonium chloride (DTAC) proved the most effective (62%, Table 1, entry 1). Importantly, no evidence of side reactions such as O–H insertion with water or C–H insertion with the surfactant was observed. All neutral surfactants performed poorly with less than 20% yield, including Lipshutz's TPGS-750-M4 (entry 2). Reactions in anionic surfactants such as dioctyl sulfosuccinate sodium salt (DOSS) or potassium laurate gave comparable results of 56% and 54%, respectively (entries 3 and 4). The control reaction in the absence of any surfactant proceeds with only a small decrease in the yield to 53% (entry 5). However, in this case, substrates 1a and 2a are both oils so it is reasonable to assume that this reaction takes place as an “on-water” reaction. To enable a more general reaction, with the improved dissolution of the starting materials, we continued using the DTAC micellar system.
All parameters of the reaction (light source, the addition of co-solvents, ratio of substrates, and surfactant concentration) were optimised leading to model cyclopropane 3a in 59% isolated yield (entry 1). Once the concentration of the surfactant exceeds 40 mM, which surpasses the CMC by ∼20 mM,33 there is a notable bump in the yield, but increasing the concentration of DTAC further has a less pronounced impact (Fig. S1, see ESI†). Only a slight increase in the yield was observed when we used the diazo compound in excess instead of styrene (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Surfactantc | Surfactant charge | Yield (%) | |
| a Standard conditions: diazo reagent 1a (0.1 mmol), styrene 2a (0.5 mmol), surfactant (0.35 mmol), H2O (5 mL), blue LEDs, and 24 h. b Diazo reagent 1a (0.5 mmol), and styrene 2a (0.1 mmol). c DTAC – dodecyltrimethylammonium chloride, TPGS-750-M – DL-α-tocopherol methoxypolyethylene glycol succinate, DOSS – dioctyl sulfosuccinate sodium salt, and SDS – sodium dodecyl sulfate. d 1H NMR yield. e GC yield. f Isolated yield. | ||||
| 1 | DTAC | Cationic | 62d (59)f | |
| 2 | TPGS-750-M | Neutral | 16e | |
| 3 | Potassium laurate | Anionic | 54e | |
| 4 | DOSS | Anionic | 56d | |
| 5 | None | N/A | 53d | |
| 6b | DTAC | Cationic | 66d | |
In a photochemical bimolecular reaction, short-lived species such as radicals and carbenes, with lifetimes in a nanoseconds range, do not diffuse out of the micelle within a period ranging from μs to ms, thus enabling a high chance of a successful collision, namely styrene in our case.34,35 As compartmentalisation has a strong impact on reactions in micellar systems, the structure (lipophilicity) of the substrates potentially has a strong impact on the process. When using diazo compounds with longer hydrophobic alkyl chains on the ester group, the yield gradually increased, from 59% for methyl (3a), to 71% for ethyl (3b), 77% for n-hexyl (3c), 82% for n-nonyl (3d) and finally 93% for n-dodecyl (3e) (Scheme 1).
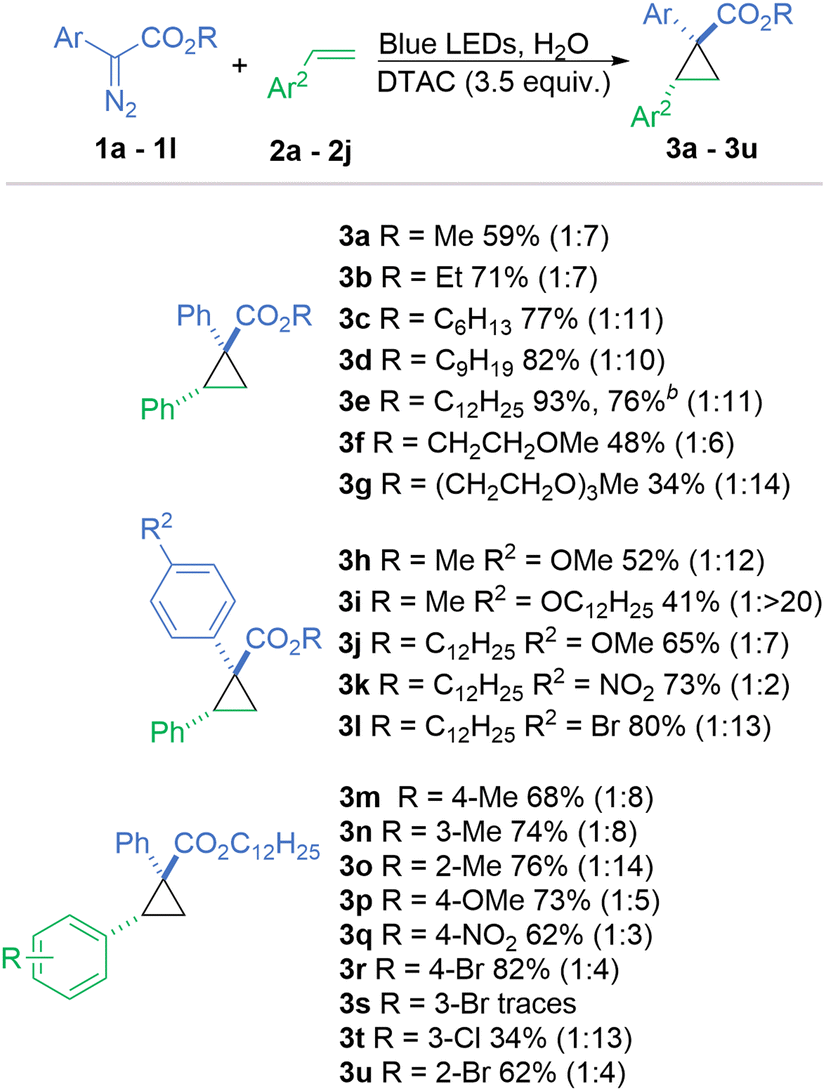 | ||
Scheme 1 Scope of the reaction. The major diastereoisomer is drawn. Reaction conditions: Diazo compounds 1a–1m (0.1 mmol), styrene 2a–2i (0.5 mmol), DTAC (0.35 mmol), H2O (5 mL), and blue LEDs. b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Diazo reagent 1e (1.0 mmol), styrene 2a (10 mmol), DTAC (3.5 mmol), H2O (50 mL), blue LEDs, and 60 h. Diazo reagent 1e (1.0 mmol), styrene 2a (10 mmol), DTAC (3.5 mmol), H2O (50 mL), blue LEDs, and 60 h. | ||
To see the impact of the diazo reagent localisation in the micelle on the reaction outcome, 1f and 1g with hydrophilic ester substituents were evaluated. The presence of oxygen atoms on their alkyl chain affects factors such as hydrophilicity, electrostatic interactions, and hydrogen bonding, which may change the position of the diazo reagent relative to styrene (2a) in the micellar compartment. In fact, reactions were less efficient compared to 3a, with cyclopropane 3f, with one glycol unit, being obtained in 48% yield with a dr of 1:6, while for cyclopropane 3g, with three glycol units, the yield dropped to 34% but had a much-improved dr of 1:14. We may therefore assume that the reduced yield for the reaction involving these diazo compounds could be because of internal non-optimal alignment in the micelles. This type of alignment is not, however, considered in COSMO-RS calculations.
Furthermore, we investigated how the position of the dodecyl chain in the diazo compound structure affects their reactivity in the micellar system. Diazo 1h, containing the 4-methoxyphenyl substituent and the methyl ester, yielded cyclopropane 3h in 52% yield. Changing the alkyl substituent on the ester from methyl to dodecyl led to a 13% increase in the yield (3i). Conversely, changing the methoxy substituent upon the aromatic ring to a dodecyloxy group led to an approximate 10% decrease in yield (3j) demonstrating that the beneficial effect from an extended alkyl chain is only relevant when it is located near to the reaction centre. Two diazo compounds 3k and 3l with electron-withdrawing aromatic substituents provided cyclopropanes in good yields although the dr is significantly eroded for product 3k possessing the nitro group.
Styrenes with electron-withdrawing and electron-donating substituents are equally effective. The substitution pattern (2-, 3- or 4-) in methyl styrenes does not influence the reaction yield in contrast to bromo styrenes (2r, 2u, and 2s), where the 3-bromo derivative only formed traces of product. Despite this, the reaction with 3-chlorostyrene (2t) could provide a serviceable amount of product 3t (34%). Expectedly, the diastereoselectivity of the reaction is influenced by the position of the substituent.
To demonstrate the applicability of the developed conditions, the reaction of diazo reagent 1e with styrene 2a was performed on a one mmol scale. Increasing the amount of styrene to 10 mmol and extending the reaction time to 60 hours enabled the construction of cycloadduct 3e in a satisfactory yield of 76% with identical dr as the 0.1 mmol scale.
Computational analysis (COSMO-RS) shows that the predicted critical micelle concentration (CMC)36 changes depending on the diazo compound used with styrene (Fig. 1, blue). The higher the predicted CMC, the higher the experimental yield. Those with hexyl, nonyl, and dodecyl chains all had a CMC of 23.5 or above and were the higher performers in our reactions. The higher the CMC, the fewer surfactant molecules are available to stabilise the micelles (DTAC = 70 mM, for all points). This would, in general, lead to larger micelles and thus more reactants are in the micellar core compared to the micelle–water interface region. The yield vs. CMC trend is negligibly changed when running the predictions when accounting for 25% product formation (Fig. 1, green). No significant trends between experimental yields and interfacial concentrations of the reactant were found, which was consistent with the reaction taking place mainly in the micellar core. No evidence of OH-insertion was observed either, which further supported our conclusion that the reaction occurred in the core.
 | ||
| Fig. 1 COSMO-RS-predicted CMC versus experimental yields. Points marked in blue represent the CMC for the reaction before any product formation (the starting point of the reaction). Points marked in green represent the CMC for the reaction after 25% product formation. | ||
Whilst our method works efficiently, there is still an inherent risk present in the work as diazo compounds are well reported for their instability and potential explosive behaviour.37 Following the work of Wu and co-workers, we explored the possibility of using bench-stable hydrazones, which can generate diazo compounds in situ with the addition of a base21 or an oxidant.38,39 Initially, we tried the reaction with a small selection of basic surfactants, namely DOSS, SDS and potassium laurate, with the idea that they could have a dual role for the reaction, as a base for the generation of the diazo compound and as a surfactant, being investigated (Scheme 2). Using DOSS or SDS only formed traces of product 3a, but we could reach a 40% yield with potassium laurate. However, DTAC in combination with a small excess of triethylamine ensured the formation of cycloadduct 3a in 59% isolated yield analogous to the reaction with the neat diazo compound and a slight increase in the diastereomeric ratio was observed too (1:7 to 1:10).
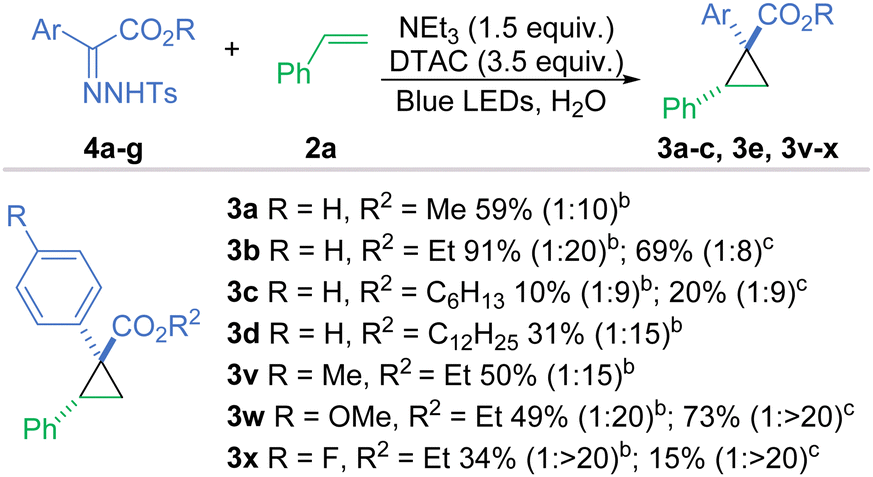 | ||
| Scheme 2 Scope of cyclopropanation with hydrazones. The major product is drawn. Conditions: Hydrazones (4a–4g) (0.1 mmol), styrene (1.0 mmol), DTAC (0.35 mmol), NEt3 (0.15 mmol), H2O (5 mL), blue LEDs, and 18–24 h. bThe hydrazone starting material was predominantly E.cThe hydrazone starting material was predominantly Z. | ||
Even though the active species, i.e. the diazo reagent, generated in the reaction is the same, we can expect some differences in the reactivity between the E- and Z-hydrazones as their stability vary and thus their propensity for deprotonation is different.40 Also, the two isomers may position themselves differently in the micelle to enable favourable electrostatic interactions and hydrogen bonding, thus influencing the rate of carbene generation.9 Thus, E- and Z-hydrazones 4b were evaluated; the E-hydrazone performed better by about 20% in terms of yield and the diastereoselectivity was significantly improved to 1:20, whereas the Z-hydrazone provided 1:8 dr. In contrast to diazo compounds, for hydrazones, increasing the length of the alkyl chain had a detrimental effect on the reaction with yields of 20% or below for hexyl esters (3c) and 31% for dodecyl ester (3e). Using hydrazones with substituents in the 4-position worked effectively and provided the products in sufficient yields. 4-Methoxy- and 4-fluoro-substituted hyrazones were highly effective for the diastereoselective synthesis of cis-cyclopropanes 3w and 3x with 1: > 20 dr.
In summary, the synthesis of cyclopropanes constitutes one of the fundamental reactions in the repertoire of carbene-derived processes and can be accomplished in water-based systems. In micellar systems, the photochemical reaction of diazo compounds with styrenes in the absence of a metal catalyst gave products in moderate to excellent yields and with good diastereoselectivity. The synthetic utility of the reaction is showcased by a one mmol scale reaction producing cyclopropane 3d in a satisfactory 76% yield. To further enhance the safety features of the reaction, the diazo compound was replaced with a hydrazone, enabling the generation of the carbene in situ by the addition of triethylamine. We believe that this work is a productive stepping stone to enable the further use of diazo compounds under photochemical conditions in the absence of organic solvents and we look forward to seeing further work in a similar vein.
J. P. M. and D. G. thank the European Union for funding under the H2020 Marie Skłodowska-Curie grant agreement No. 956324 (MSCA ITN: PhotoReAct). D. G. thanks the National Science Centre, Poland grant number Maestro UMO-2020-38/A/ST4/00185 for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Gallou, N. A. Isley, A. Ganic, U. Onken and M. Parmentier, Green Chem., 2015, 18, 14–19 RSC.
- M. Billamboz, F. Mangin, N. Drillaud, C. Chevrin-Villette, E. Banaszak-Léonard and C. Len, J. Org. Chem., 2014, 79, 493–500 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- C. Russo, F. Brunelli, G. C. Tron and M. Giustiniano, J. Org. Chem., 2023, 88, 6284–6293 CrossRef CAS PubMed.
- C. Chatgilialoglu, S. Barata-Vallejo and T. Gimisis, Molecules, 2024, 29, 569 CrossRef CAS PubMed.
- G. Pölderl and L. Næsborg, ChemPhotoChem, 2024, e2023003 Search PubMed.
- B. H. Lipshutz, Green Chem., 2023, 26, 739–752 RSC.
- L. Brüss, R. Jeyaseelan, J. C. G. Kürschner, M. Utikal and L. Næsborg, ChemCatChem, 2022, 15, e202201146 CrossRef.
- M. Banerjee, P. C. Panjikar, Z. T. Bhutia, A. A. Bhosle and A. Chatterjee, Tetrahedron, 2021, 88, 132142 CrossRef CAS.
- G. La Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644–683 RSC.
- E. Borrego, A. Caballero and P. J. Pérez, Organometallics, 2022, 41, 3084–3098 CrossRef CAS PubMed.
- H. K. Maurya, Org. Chem. Ind. J., 2022, 16, 1 Search PubMed.
- A. Steven, Synthesis, 2019, 2632–2647 CAS.
- M. Banerjee, P. C. Panjikar, Z. T. Bhutia, A. A. Bhosle and A. Chatterjee, Tetrahedron, 2021, 88, 132142 CrossRef CAS.
- M. Cybularczyk-Cecotka, J. Predygier, S. Crespi, J. Szczepanik and M. Giedyk, ACS Catal., 2022, 12, 3543–3549 CrossRef CAS.
- R. Jeyaseelan, M. Utikal and L. Næsborg, Asian J. Org. Chem., 2024, e202300604 CrossRef CAS.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed.
- J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS.
- S. Vidal, ACS Cent. Sci., 2020, 6, 83–86 CrossRef CAS PubMed.
- M. Tobiszewski, J. Namieśnik and F. Pena-Pereira, Green Chem., 2017, 19, 1034–1042 RSC.
- I. Nicolas, P. Le Maux and G. Simonneaux, Coord. Chem. Rev., 2008, 252, 727–735 CrossRef CAS.
- M. Álvarez, R. Gava, M. R. Rodríguez, S. G. Rull and P. J. Pérez, ACS Catal., 2017, 7, 3707–3711 CrossRef.
- C.-M. Ho, J.-L. Zhang, C.-Y. Zhou, O.-Y. Chan, J. J. Yan, F.-Y. Zhang, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2010, 132, 1886–1894 CrossRef CAS PubMed.
- N. R. Candeias, C. Carias, L. F. R. Gomes, V. André, M. T. Duarte, P. M. P. Gois and C. A. M. Afonso, Adv. Synth. Catal., 2012, 354, 2921–2927 CrossRef CAS.
- N. R. Candeias, P. M. P. Gois and C. A. M. Afonso, J. Org. Chem., 2006, 71, 5489–5497 CrossRef CAS PubMed.
- N. R. Candeias, P. M. P. Gois and C. A. M. Afonso, Chem. Commun., 2005, 391–393 RSC.
- Q. Z. Li, X. Zhang, K. Xie, Q. S. Dai, R. Zeng, Y. Q. Liu, Z. Q. Jia, X. Feng and J. L. Li, Green Chem., 2019, 21, 2375–2379 RSC.
- N. R. Candeias, P. M. P. Gois, L. F. Veiros and C. A. M. Afonso, J. Org. Chem., 2008, 73, 5926–5932 CrossRef CAS PubMed.
- K. Yan, H. He, J. Li, Y. Luo, R. Lai, L. Guo and Y. Wu, Chin. Chem. Lett., 2021, 32, 3984–3987 CrossRef CAS.
- R. V. Maaskant, E. A. Polanco, R. C. W. Van Lier and G. Roelfes, Org. Biomol. Chem., 2020, 18, 638–641 RSC.
- S. K. Mehta, K. K. Bhasin, R. Chauhan and S. Dham, Colloids Surf., A, 2005, 255, 153–157 CrossRef CAS.
- N. J. Turro, M. F. Chow, C. J. Chung, Y. Tanimoto and G. C. Weed, J. Am. Chem. Soc., 1981, 103, 4574–4576 CrossRef CAS.
- N. J. Turro, G. Sidney Cox and M. A. Paczkowski, Photochemistry in micelles, Springer Berlin Heidelberg, Berlin, Heidelberg, 1985 Search PubMed.
- M. Turchi, A. P. Karcz and M. P. Andersson, J. Colloid Interface Sci., 2022, 618–627 CrossRef CAS PubMed.
- S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 24, 67–84 CrossRef CAS PubMed.
- W. Liu, J. Twilton, B. Wei, M. Lee, M. N. Hopkins, J. Bacsa, S. S. Stahl and H. M. L. Davies, ACS Catal., 2021, 11, 2676–2683 CrossRef CAS.
- N. Tanbouza, L. Caron, A. Khoshoei and T. Ollevier, Org. Lett., 2022, 24, 2675–2678 CrossRef CAS PubMed.
- P. Tisovský, J. Donovalová, R. Sokolík, M. Horváth and A. Gáplovský, ChemistrySelect, 2021, 6, 10651–10654 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, compound characterisation (NMR, HRMS), and NMR spectra. See DOI: https://doi.org/10.1039/d4cc00828f |
| This journal is © The Royal Society of Chemistry 2024 |