
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic enantioselective oxa-Piancatelli rearrangement†
Rahul
Sarkar
,
Alexander
Korell
and
Christoph
Schneider
 *
*
Institut für Organische Chemie, Universität Leipzig, Leipzig D-04103, Germany. E-mail: schneider@chemie.uni-leipzig.de; Fax: +49 341 97-36559; Tel: +49 341 97-36559
First published on 19th February 2024
Abstract
The first highly enantioselective oxa-Piancatelli rearrangement has been developed. This process which is catalyzed by a chiral BINOL-derived phosphoric acid rearranges a wide range of furylcarbinols into densely substituted γ-hydroxy cyclopentenones in high yield with excellent diastereo- and enantioselectivities (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er). This reaction exhibits a high functional group tolerance and was applied to complex bioactive molecules as well. The products were further manipulated into value-added molecular scaffolds further highlighting their versatility and synthetic utility.
:1 er). This reaction exhibits a high functional group tolerance and was applied to complex bioactive molecules as well. The products were further manipulated into value-added molecular scaffolds further highlighting their versatility and synthetic utility.
The Piancatelli rearrangement, a unique process that directly transforms furylcarbinols into functionalized cyclopentenones, was first discovered by Piancatelli in 1976.1 This reaction proceeds through an acid catalyzed dehydration of the furyl-2-carbinol to generate a highly reactive furanoxonium ion, which suffers nucleophilic attack of H2O and undergoes ring-opening to form a pentadienyl cation. Subsequent conrotatory 4π-electrocyclization provides γ-hydroxy cyclopentenones with trans diastereoselectivity (Scheme 1A).2 The products are important molecular scaffolds, which are not only present in several natural products,3 but also serve as precursors for the synthesis of various bioactive compounds and naturally occurring molecules such as prostaglandin derivatives,3a,4 sibirinone,5 and verrillin.6
 | ||
| Scheme 1 Catalytic Piancatelli rearrangement. | ||
Significant advancements in the development of new catalytic systems, including those based on Lewis acids, have occurred over the past two decades and led to the invention of a large family of catalytic transformations involving various internal and external O-, N- and C-nucleophiles, albeit only in racemic fashion.7 Rueping, Sun, and Patil then independently reported the first catalytic, enantioselective processes, namely chiral Brønsted acid catalyzed enantioselective aza-Piancatelli rearrangements (Scheme 1B).8 For the original oxa-Piancatelli rearrangement, however, only a single enantioselective process has been reported to date with a chiral vanadium complex which delivers a small selection of products with generally moderate enantioselectivity.9
Major challenges associated with the asymmetric oxa-Piancatelli rearrangement are the attenuated nucleophilicity of H2O in comparison to anilines10 and an undesired isomerization of the products yielding difficult to separate mixtures.11,12 More importantly, in comparison to the aza-Piancatelli variant, the key step of ring-opening the hemiacetal to the pentadienyl cation is significantly retarded due to the reduced resonance effect of the hydroxyl group.12 Considering the importance of densely substituted and enantiomerically enriched γ-hydroxy cyclopentenones, the development of an efficient, highly enantioselective process is highly desirable.13
We herein report the BINOL phosphoric acid-catalyzed, highly enantioselective oxa-Piancatelli rearrangement (Scheme 1C). We envisioned that introduction of an electron-donating group at the C3 position of the furyl-2-carbinol would not only increase the stability of the in situ generated furanoxonium ion, but more importantly aid in the ring-opening of the hemiacetal into the pentadienyl cation through its resonance effect. The catalyst-induced 4π-electrocyclic ring closure would eventually provide the desired γ-hydroxy cyclopentenones in enantiomerically highly enriched form (Scheme 1C).
Accordingly, we began our investigations with the optimization of the catalyst and reaction conditions for p-methoxyphenyl (PMP) substituted secondary furyl-2-carbinol 1a and tertiary furyl-2-carbinol 2a in dichloromethane at 25 °C. In the presence of 10 mol% (R)-BINOL-derived phosphoric acid 3a as catalyst and H2O (2.0 equiv.) as an additive, the reaction of secondary alcohol 1a proceeded in the expected fashion to generate the γ-hydroxy cyclopentenone 4a with promising enantioselectivity, albeit in only a 44% yield. Similarly, tertiary alcohol 2a also participated in the reaction under the same reaction conditions, providing the product 5a in moderate yield with good enantioselectivity (Scheme 2). Screening of various BINOL phosphoric acids 3b–3g and different solvents revealed that catalyst 3g in a 1.5:1 mixture of toluene and dichloromethane as the solvent was optimal for the reaction and afforded the product 4a in 96% isolated yield with 98:2 er (for further details, see the ESI†). In addition, the conditions optimized for 1a were equally effective for tertiary alcohol 2a, providing the product 5a in 93% isolated yield with 99:1 er (Scheme 2, bottom).
 | ||
| Scheme 2 Optimization of the reaction conditions. | ||
After optimizing the catalyst and reaction conditions for 1a and 2a (Scheme 2), we sought to test scope and limitations of this novel process (Table 1). Overall, it is quite generally applicable, and furyl-2-carbinols (1a–1n) bearing either electron-donating or electron-withdrawing substituents at various positions of the aryl ring at the carbinol center smoothly underwent the reaction to provide the corresponding rearranged products 4a–4n as single diastereomers with high yields and excellent enantioselectivities (Table 1A). The relative and absolute configuration of 4j was determined by single crystal X-ray diffraction analysis, and those of other products were assigned by analogy (CCDC 2295678; Table 1).14 Apart from simple aryl substituents, alcohols containing polyaromatic hydrocarbons 1o–1q also effectively participated in the reaction and generated the products 4o–4q in good yields with high er (Table 1B). Moreover, we successfully incorporated pharmaceutically relevant heterocycles into our products. For example, cyclopentenones carrying a 3-thiophenyl (4r), 2-thiophenyl (4s), and dioxolane substituent (4t) were obtained in moderate to good yields with excellent enantioselectivity (Table 1C). As observed by Piancatelli and coworkers, alkyl- and cycloalkyl-substituted furyl-2-carbinols are generally more resistant towards an acid catalyzed rearrangement. Thus, they typically require more drastic reaction conditions to effect the oxa-Piancatelli rearrangement or do not rearrange at all.15 To our great delight, both alkyl- and cycloalkyl substituted furyl-2-carbinols efficiently reacted under the optimized reaction conditions (Table 1D). For example, γ-hydroxy cyclopentenones containing linear (4u and 4v) and branched alkyl groups (4w and 4x) as well as homoallyl (4y) and homobenzyl (4z) groups were isolated with good yields and excellent enantioselectivities. Additionally, cycloalkyl substituted furyl-2-carbinols successfully participated in this reaction and gave rise to products 4aa and 4ab in moderate to good yields and with high enantioselectivity again. The yield of these reactions was slightly reduced compared to those with aryl-substituted carbinols likely based upon the attenuated resonance stabilization of the cationic intermediate. The effect of the C3-substituent on the rearrangement was also briefly investigated (Table 1E). The PMP-group could be easily replaced with a phenyl (4ac), p- and o-tolyl (4ad and 4ae), and p- and o-fluorophenyl groups (4af and 4ag) without compromising either yield or enantioselectivity. 1-Naphthyl and thiophenyl-substituted cyclopentenones 4ah and 4ai, however, were obtained with somewhat diminished er.
|
a Reaction conditions: 0.1 mmol of 1, 0.2 mmol of H2O, and catalyst 3g (10 mol%) in 1.0 mL toluene/CH2Cl2 (1.5:1). The diastereomeric ratio (dr) was determined by 1H NMR of the crude reaction mixture and was >20:1 in all cases. Yields correspond to the isolated product after chromatographic purification. The er was determined by HPLC analysis on a chiral stationary phase.
|
|---|

|
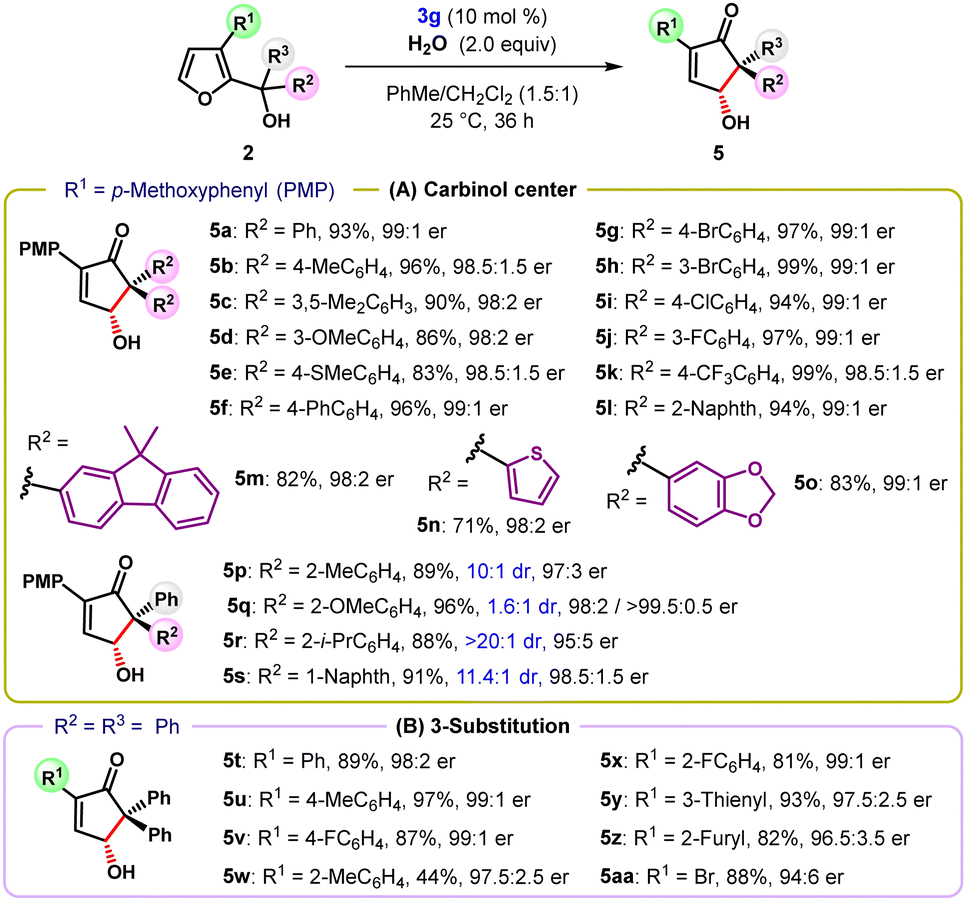
The oxa-Piancatelli rearrangement of tertiary furyl-2-carbinols was studied next (Table 2). A diverse array of tertiary carbinols carrying either electron-rich (e.g., Me, OMe, SMe, and Ph) or electron-withdrawing (e.g., Br, Cl, F, and CF3) groups at the meta- and para-positions of the aryl ring at the carbinol center were well tolerated and provided the products 5a–5k in uniformly high yield and with excellent enantioselectivity (Table 2A). In addition to aryl substituents, substrates 5l–5o with polyaromatic and heterocyclic groups underwent this reaction with equal efficacy. ortho-Substituted diaryl alcohols failed to deliver the desired products, possibly due to severe steric congestion in the transient planar pentadienyl cation. However, unsymmetrically substituted alcohol 2p with o-tolyl and phenyl groups at the carbinol center delivered cyclopentenone 5p with a quaternary stereocenter in high yield and with 10:1 dr and 97:3 er. Likewise, tertiary furylcarbinols bearing o-isopropylphenyl and 1-naphthyl substituents furnished products 5r and 5s with excellent yields and high diastereo- and enantioselectivity. In accordance with previous reports,15c tertiary furylcarbinols containing an alkyl substituent at the carbinol center were readily dehydrated to yield stable alkenes as the major products.16 Next, the scope of the reaction was extended to tertiary furyl-2-carbinols carrying different substituents at the C3 position in place of the PMP-group (Table 2B). A range of other 2-aryl-substituted γ-hydroxy cyclopentenones 5t–5x were obtained with generally high enantiocontrol, irrespective of the electronic nature of the aryl group. Moreover, heteroaryl residues (5y and 5z) were equally effective for promoting the oxa-Piancatelli rearrangement. Intriguingly, a simple bromine substituent capable of releasing electron density into the furan ring also enabled the reaction affording the product 5aa in high yield and with good enantioselectivity.
|
a Reaction conditions: 0.1 mmol of 2, 0.2 mmol of H2O, and catalyst 3g (10 mol%) in 1.0 mL toluene/CH2Cl2 (1.5:1). The dr was determined by 1H NMR of the crude reaction mixture. Yields correspond to the isolated product after chromatographic purification. The er was determined by HPLC analysis on a chiral stationary phase.
|
|---|
|
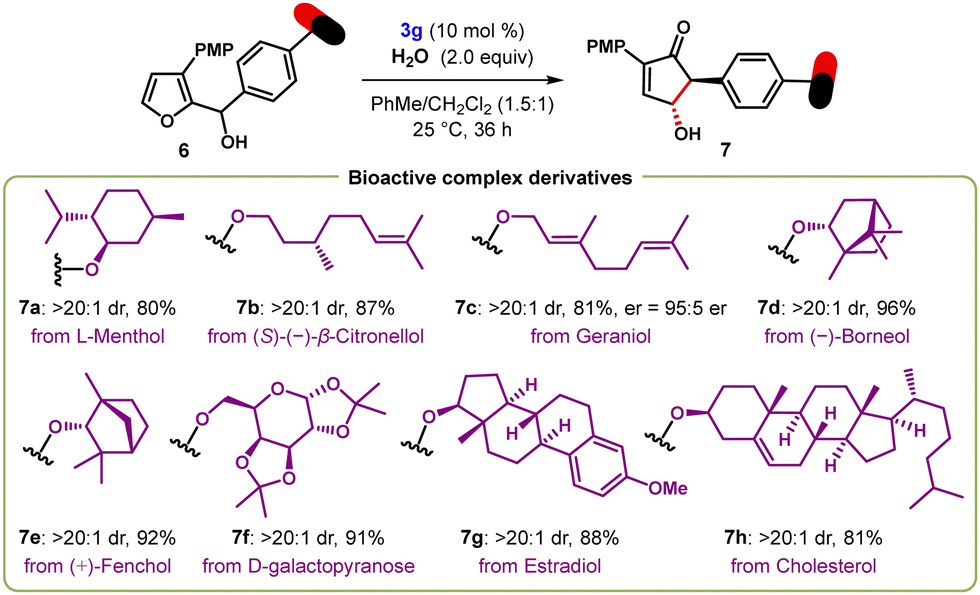
The mild reaction conditions of this transformation and the efficiency displayed by secondary furyl-2-carbinols 1a–1ai allowed the use of substrates with complex bioactive residues showcasing the functional group compatibility of this process (Scheme 3). For example, furylcarbinols derived from L-menthol 6a, (S)-β-citronellol 6b, and geraniol 6c smoothly participated in the oxa-Piancatelli rearrangement to generate the products 7a–7c in high yields and with excellent diastereoselectivities (20:1 dr). Similarly, the rearrangement of 6d and 6e derived from (−)-borneol and (+)-fenchol, respectively, occurred efficiently to produce the corresponding cyclopentenones 7d and 7e with excellent yield and selectivity. D-Galactopyranose-derived alcohol 6f was successfully transformed into 7f with high diastereocontrol (>20:1 dr). Notably, such as estradiol 6g and cholesterol 6h, also reacted in the expected fashion to give rise to the products 7g–7h as single diastereomers in high yields.
 | ||
| Scheme 3 Substrates with complex bioactive residues. | ||
The scalability of our protocol was demonstrated by performing the reaction of 1a on a 2.0 mmol scale (Scheme 4A). Under the optimal reaction conditions, product 4a was obtained in 94% yield with the same level of enantiopurity as in the smaller scale reaction.
 | ||
| Scheme 4 (A) Scale-up reaction and synthetic elaborations of γ-hydroxycyclopentenones. (B) Control experiments. | ||
To show the synthetic utility of this process, the enantioenriched γ-hydroxy cyclopentenones were converted into synthetically useful building blocks (Scheme 4A). Toward this goal, the free hydroxy group of 4a was protected with the TBS group to obtain the silyl ether 8 in 88% yield. Reduction of 8 with LiBH4 in the presence of CeCl3·7H2O was completely regio- and diastereoselective and furnished the alcohol 9 in 64% yield. Treatment of 8 with vinylmagnesium bromide resulted in the formation of tertiary alcohol 10 in 63% yield, albeit with only 1.2:1 dr. However, both diastereomers of 10 were separable through chromatographic purification. In all cases, the reactions proceeded with only minimal or no deterioration of enantiopurity. TBS-protection of 5a using TBSOTf afforded the silyl ether 11 in 97% yield. Selective hydrogenation of the electron-deficient olefin of 11 was achieved with catalytic Pd/C, and the resulting poly-substituted cyclopentanone 12 was obtained as a single diastereomer in 76% yield.
Two control experiments with C3-unsubstituted furyl-2-carbinols were conducted under otherwise identical reaction conditions: while the secondary furyl-2-carbinol (R1 = H, R2 = Ph) only gave rise to decomposition of the substrate, the tertiary furyl-2-carbinol 2′ (R1 = H, R2, R3 = Ph) did produce the desired Piancatelli product 5′, albeit in only 16% yield. Again, significant decomposition of starting material was observed leading to 11% of benzophenone as the only isolable side product (Scheme 4B, eqn (i)). Furthermore, shifting the p-methoxy-phenyl (PMP) group from C3 to the C4 position within the furan nucleus of 1a prevented the PMP group from stabilizing the furanoxonium ion through its resonance effect. As a result, carbinol 1a′ carrying a PMP group at the C4 position failed to produce any product under standard conditions (Scheme 4B, eqn (ii)). These control experiments corroborate our initial mechanistic assumption about the role of the resonance-stabilizing C3-substituent.
In conclusion, we have developed the first catalytic, highly enantioselective oxa-Piancatelli reaction. This one-step and operationally simple process is catalyzed by a chiral BINOL-derived phosphoric acid and converts a wide range of easily accessible furyl-2-carbinols into highly valuable, substituted γ-hydroxy cyclopentenones with typically high yields and excellent levels of diastereo- and enantioselectivity. The synthetic potential of this reaction is apparent in the light of the prostaglandin class of natural products and was further demonstrated by transforming the products into other densely functionalized, chiral structural motifs.
This work was generously supported by the Deutsche Forschungsgemeinschaft (SCHN 441/16-1). We are grateful to Dr Peter Lönnecke (University of Leipzig) for the X-ray structure analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Piancatelli, A. Scettri and S. Barbadoro, Tetrahedron Lett., 1976, 17, 3555–3558 CrossRef.
- (a) M. J. Palframan and G. Pattenden, Chem. Commun., 2014, 50, 7223–7242 RSC; (b) C. Piutti and F. Quartieri, Molecules, 2013, 18, 12290–12312 CrossRef CAS PubMed; (c) O. Nieto Faza, C. Silva López, R. Álvarez and Á. R. de Lera, Chem. – Eur. J., 2004, 10, 4324–4333 CrossRef CAS PubMed.
- (a) S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744–5893 CrossRef CAS PubMed; (b) S. P. Roche and D. J. Aitken, Eur. J. Org. Chem., 2010, 5339–5358 CrossRef CAS.
- (a) R. Katsuta, K. Aoki, A. Yajima and T. Nukada, Tetrahedron Lett., 2013, 54, 347–350 CrossRef CAS; (b) J. P. Henschke, Y. Liu, X. Huang, Y. Chen, D. Meng, L. Xia, X. Wei, A. Xie, D. Li, Q. Huang, T. Sun, J. Wang, X. Gu, X. Huang, L. Wang, J. Xiao and S. Qiu, Org. Process Res. Dev., 2012, 16, 1905–1916 CrossRef CAS; (c) A. R. Rodríguez and B. W. Spur, Tetrahedron Lett., 2003, 44, 7411–7415 CrossRef; (d) A. Rodríguez, M. Nomen, B. W. Spur and J.-J. Godfroid, Eur. J. Org. Chem., 1999, 2655–2662 CrossRef; (e) J. H. Dygos, J. P. Adamek, K. A. Babiak, J. R. Behling, J. R. Medich, J. S. Ng and J. J. Wieczorek, J. Org. Chem., 1991, 56, 2549–2552 CrossRef CAS; (f) G. Stork, C. Kowalski and G. Garcia, J. Am. Chem. Soc., 1975, 97, 3258–3260 CrossRef CAS PubMed.
- D. Dobler and O. Reiser, J. Org. Chem., 2016, 81, 10357–10365 CrossRef CAS PubMed.
- A. Saitman and E. A. Theodorakis, Org. Lett., 2013, 15, 2410–2413 CrossRef CAS PubMed.
- Recent reviews: (a) S. Zhong, L. Xu and Y. Cai, Synthesis, 2022, 589–599 CAS; (b) C. Verrier, S. Moebs-Sanchez, Y. Queneau and F. Popowycz, Org. Biomol. Chem., 2018, 16, 676–687 RSC; (c) R. F. A. Gomes, J. A. S. Coelho and C. A. M. Afonso, Chem. – Eur. J., 2018, 24, 9170–9186 CrossRef CAS PubMed ; selected recent articles: ; (d) P. R. Solanke, R. Cinsani, K. Nayani, P. S. Mainkar and S. Chandrasekhar, Chem. Commun., 2022, 58, 5530–5533 RSC; (e) F. Cacheux, G. Le Goff, J. Ouazzani, J. Bignon, P. Retailleau, A. Marinetti, A. Voituriez and J.-F. Betzer, Org. Chem. Front., 2021, 8, 2449–2455 RSC; (f) S. Wang, R. Guillot, J.-F. Carpentier, Y. Sarazin, C. Bour, V. Gandon and D. Leboeuf, Angew. Chem., Int. Ed., 2020, 59, 1134–1138 CrossRef CAS PubMed; (g) D. Leboeuf, L. Marin, B. Michelet, A. Perez-Luna, R. Guillot, E. Schulz and V. Gandon, Chem. – Eur. J., 2016, 22, 16165–16171 CrossRef CAS PubMed; (h) D. Fisher, L. I. Palmer, J. E. Cook, J. E. Davis and J. Read de Alaniz, Tetrahedron, 2014, 70, 4105–4110 CrossRef CAS; (i) L. I. Palmer and J. Read de Alaniz, Angew. Chem., Int. Ed., 2011, 50, 7167–7170 CrossRef CAS PubMed.
- (a) Y. Cai, Y. Tang, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 14126–14130 CrossRef CAS PubMed; (b) H. Li, R. Tong and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 15125–15128 CrossRef CAS PubMed; (c) A. B. Gade and N. T. Patil, Synlett, 2017, 1096–1100 CAS for aza-Piancatelli rearrangements embedded into domino processes see: ; (d) B. Shen, Q. He, S. Dong, X. Liu and X. Feng, Chem. Sci., 2020, 11, 3862–3867 RSC; (e) L. Xu, Q. Yang, S. Zhong, H. Li, Y. Tang and Y. Cai, Org. Lett., 2020, 22, 9016–9021 CrossRef CAS PubMed.
- L. Schober, M. Sako, S. Takizawa, H. Gröger and H. Sasai, Chem. Commun., 2020, 56, 10151–10154 RSC.
- (a) T. Hashimoto, Y. Shimazaki, Y. Omatsu and K. Maruoka, Angew. Chem., Int. Ed., 2018, 57, 7200–7204 CrossRef CAS PubMed; (b) R. M. Demming, S. C. Hammer, B. M. Nestl, S. Gergel, S. Fademrecht, J. Pleiss and B. Hauer, Angew. Chem., Int. Ed., 2019, 58, 173–177 CrossRef CAS PubMed.
- (a) A. Scettri, G. Piancatelli, M. D'Auria and G. David, Tetrahedron, 1979, 35, 135–138 CrossRef CAS; (b) G. Piancatelli and A. Scettri, Synthesis, 1977, 116–117 CrossRef CAS.
- See the ESI† Section III for details.
- These studies were inspired by our recent endeavors in ortho-quinone methide chemistry. For a recent review see: C. Dorsch and C. Schneider, Synthesis, 2022, 3125–3141 CAS.
- CCDC 2295678† contains the crystallographic data for 4j.
- (a) G. Piancatelli, M. D’Auria and F. D’Onofrio, Synthesis, 1994, 867–889 CrossRef CAS; (b) G. Piancatelli and A. Scettri, Tetrahedron, 1977, 33, 69–72 CrossRef CAS; (c) G. Piancatelli and A. Scettri, Tetrahedron Lett., 1977, 18, 1131–1134 CrossRef.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and analytical data. CCDC 2295678. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc00708e |
| This journal is © The Royal Society of Chemistry 2024 |