
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective encapsulation of carboxylic acid dimers within a size-regulable resorcinarene-based hemicarcerand†
Kentaro
Harada
a,
Yudai
Ono
 ab,
Ryo
Sekiya
a and
Takeharu
Haino
*ab
ab,
Ryo
Sekiya
a and
Takeharu
Haino
*ab
aDepartment of Chemistry, Graduate School of Advanced Science and Engineering Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima, Hiroshima, 739-8526, Japan. E-mail: haino@hiroshima-u.ac.jp
bInternational Institute for Sustainability with Knotted Chiral Meta Matter (WPI-SKCM2), Hiroshima University, 2-313 Kagamiyama, Higashi-Hiroshima, Hiroshima, 739-0046, Japan
First published on 31st May 2024
Abstract
A cavity within a resorcinarene-based hemicarcerand was contracted and expanded through conformational changes induced by the complexation and decomplexation, allowing self-sorting of homo- and heterodimeric carboxylic acid pairs.
Allosteric regulation is crucial for controlling metabolic pathways, which modify the activity of proteins in response to effector binding.1–5 For example, calmodulin exhibits allosteric behavior.6 When a calcium ion binds to the remote site of calmodulin, the structure of the hydrophobic binding pocket is deformed. This process allows for the selective binding of target proteins, such as myosin light-chain kinase. Abiotic molecular capsules with large internal cavities that can accommodate guests with large molecular dimensions have been developed to mimic the allosteric regulation of proteins.7–25 Light,26–31 metals,32–35 pH,36–39 and anions40,41 act as effectors that activate or deactivate binding sites, which drive the uptake or release of guest molecules in an allosteric manner. Abiotic allosteric molecules can be applied in many potential applications, including drug delivery,42,43 catalysis,44,45 and sensing.46,47 Many efforts have been devoted to developing size-adjustable abiotic capsules in which guest encapsulation is switchable however, developing an artificial molecular cavity that can self-sort specific guest pairs among several possible pairs has been a great scientific challenge.48–53
Hemicarcerand 1 is a unique molecular container that possesses an internal cavity (Fig. 1a). This cavity can be contracted and expanded by metal complexation and decomplexation, respectively, which is accompanied by switchable molecular recognition.54,55 Herein, we report the development of homo and heterodimeric carboxylic acid pairs with self-sorting behaviors inside the cavity of 1. Carboxylic acids are known to dimerize in lipophilic solvents,56,57 and mixtures of multiple carboxylic acids generate homo and heterodimeric pairs according to a statistical distribution. Thus, it is generally difficult to selectively obtain a desired dimeric pair. The cavity of 1 and the Ag-coordinated hemicarcerand (1Ag) provided expanded and contracted environments, respectively, in which homo (G2a·G2a) or heterodimeric (G1a·G2a) pairs were selectively formed in a self-sorting manner (Fig. 1b). The selective encapsulation of the dimeric carboxylic acid pairs was reversibly regulated upon the addition and removal of Ag+ cations.
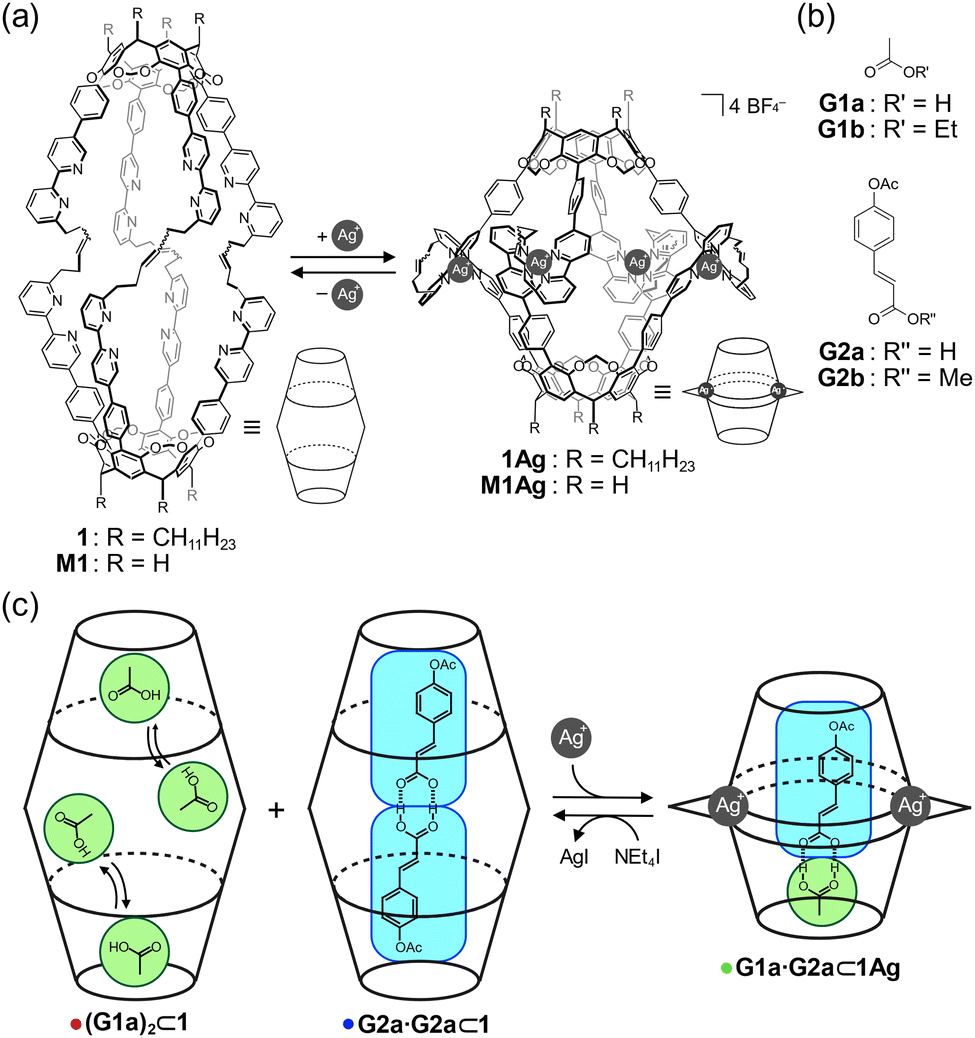 | ||
| Fig. 1 (a) Molecular structures of 1 and 1Ag and those of M1 and M1Ag for DFT calculations. (b) Guests G1a–b and G2a–b. (c) Schematic representation showing the self-sorting behavior of homo and heterodimeric carboxylic acid pairs. | ||
Reversible contraction and expansion of the cavity was achieved by the interconversion between 1 and 1Ag. The aromatic proton signals of 1 were assigned entirely in chloroform-d1 (Fig. 2a). The addition of two equivalents of AgBF4 to a solution of 1 caused downfield shifts in the pyridyl protons Ha–He as a result of the reduced electron density of the bipyridyl arms due to Ag–N coordination. In contrast, the pyridyl proton Hf, which was located within the shielding region of the neighboring bipyridyl arm in the tetrahedral coordination geometry, exhibited an upfield shift of 0.50 ppm. The eight arms showed equivalent signals, indicative of the D4 symmetry on the NMR time scale. The electrospray ionization mass spectrum showed ion peaks corresponding to [1+Ag4]4+ and [1+Ag4(BF4)]3+. The isotope pattern of [1+Ag4(BF4)]3+ corresponded well with the simulated result (Fig. S3 in ESI†).
 | ||
| Fig. 2 Selected region of the 1H NMR spectra (500 MHz, chloroform-d1, 298 K) of (a) 1 and (b) 1Ag. | ||
The host–guest complexation of 1 was evaluated by 1H NMR spectroscopy (Fig. 3). A mixture of 1 and G1a generated methyl protons of bound G1a at approximately δ = −1.6 ppm (Fig. 3a), which led to exchange cross peaks between the bound and free G1a (Fig. S9 in ESI†). The large upfield shifts indicated that G1a was encapsulated in 1. The signals consisted of broad and weak sharp signals. As the bipyridyl arms function as Lewis bases, G1a can anchor to the bipyridyl arms. Thus, the methyl protons of bound guests show sharp signals due to their methyl groups at the bottom of the capsules, which form C–H/π interactions.54,58 Hence, the sharp signal was tentatively assigned as G1a at the bottom of 1, and the broad signal originated from G1a anchoring to the bipyridyl arms. The integration of the signals indicated that two molecules of G1a were encapsulated in 1 (Fig. S4 in ESI†). This host–guest complex was named (G1a)2⊂1.
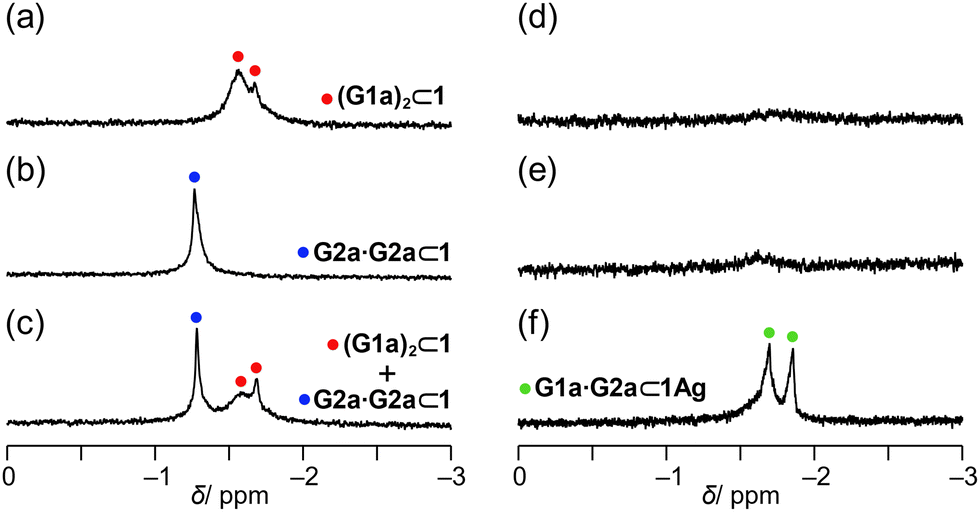 | ||
| Fig. 3 Selected region of the 1H NMR spectra (500 MHz, chloroform-d1, 223 K) of the mixture of capsule 1 (1.5 mM) and (a) G1a (15 mM), (b) G2a (15 mM), and (c) G1a (15 mM) and G2a (15 mM) and the mixture of capsule 1Ag (1.5 mM) and (d) G1a (15 mM), (e) G2a (15 mM), and (f) G1a (15 mM) and G2a (15 mM). | ||
A mixture of 1 and G2a generated only a sharp signal corresponding to the methyl protons of G2a at δ = −1.27 ppm (Fig. 3b). Based on the signal intensity, the host–guest ratio was determined to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Fig. S12 in the ESI†). NOEs were observed between the methyl protons of G2a and the methylene bridges of 1 (Fig. S11 in ESI†), indicating that the methyl group was located at the bottom of 1, resulting in the C–H/π interactions. Hence, the carboxy group was pointed to the center of the cavity, indicating that the hydrogen-bonded dimer was formed in the host–guest complex G2a·G2a⊂1.
:2 (Fig. S12 in the ESI†). NOEs were observed between the methyl protons of G2a and the methylene bridges of 1 (Fig. S11 in ESI†), indicating that the methyl group was located at the bottom of 1, resulting in the C–H/π interactions. Hence, the carboxy group was pointed to the center of the cavity, indicating that the hydrogen-bonded dimer was formed in the host–guest complex G2a·G2a⊂1.
A control experiment showed that the COOH group is required for the host–guest complexation of 1 when esters G1b and G2b were employed. 1H NMR spectra showed no signals assignable to bound G1b and G2b (Fig. S13 in ESI†). Because 1 can adjust its cavity for the bound guests,54 the longer size of G2b than G2a would not be responsible for the no host–guest complexation. Further, in THF-d8G2a was not trapped in 1 (Fig. S23 in ESI†). THF functions as a hydrogen bond acceptor. Thus, the hydrogen-bonded dimeric pair of G2a is weakened by the competitive solvation, which most likely interferes with host–guest complexation in THF-d8, although the possibility of the encapsulation of THF in 1 instead of G2a cannot be ruled out. In view of these facts, the hydrogen-bonded dimeric form of G2a is crucial for host–guest complexation.
When G1a and G2a were mixed with 1, two sets of signals appeared, which were assignable to (G1a)2⊂1 and G2a·G2a⊂1 (Fig. 3c). The other signals of bound G2a·G2a resonated at the same chemical shifts as those of G2a·G2a⊂1 alone (Fig. S4 in ESI†). When the heterodimeric form was organized, the methyl protons of G1a and G2a were expected to generate a 1:1 signal ratio, as in the case of 1Ag (see below). However, these signals were not detected. These observations rationalize that the homodimeric form of G2a or two molecules of G1a were selectively encapsulated in 1.
1Ag showed a distinct selectivity. Although a mixture of 1Ag with G1a or G2a generated very weak signals (Fig. 3d and e), a mixture of 1Ag with G1a and G2a generated methyl protons of G2a and G1a with a 1:1 signal intensity at δ = −1.69 ppm and −1.86 ppm, respectively. Exchange peaks were observed between bound and unbound G1a and G2a (Fig. 4a). The methyl protons of bound G1a and G2a generated NOE correlations with the bridge methylene signals Hj at 223 K (Fig. 4b), revealing that the methyl groups of G1a and G2a were located at the bottom of 1Ag; thus, the carboxy groups of these compounds faced each other in the cavity. Accordingly, G1a·G2a was likely to be organized in 1Ag. The host–guest complexation was supported by 1H DOSY, wherein the diffusion coefficients of bound G1a and G2a were consistent with that of 1Ag (Fig. 4c).
 | ||
| Fig. 4 (a) and (b) Selected region of the 2D NOESY spectra (500 MHz, chloroform-d1) of the mixture of 1Ag (1.5 mM), G1a (15 mM) and G2a (15 mM) at (a) 298 K and (b) 223 K. (c) DOSY spectra (500 MHz, chloroform-d1, 298 K) of the mixture of 1Ag (1.5 mM), G1a (15 mM) and G2a (15 mM). The blue and green filled circles denote bound G2a and G1a, respectively, and the blue filled triangles denote unbound G2a. | ||
To evaluate the structural feasibility of the host–guest complexes, density functional theory calculations were carried out using M1 and M1Ag, in which the long alkyl chains on the lower rim were replaced with hydrogen atoms.59 The optimized structures of G1a·G2a⊂M1 (Fig. S22a in ESI†) and G2a·G2a⊂M1 (Fig. 5a) suggested that the homodimer fit well within M1. For G1a·G2a⊂M1, the cavity must be squeezed to fit the heterodimer, which involves folding the alkyl chains that connect two cavitands; these movements cause an energetic penalty, which should contribute to the selective organization of G2a·G2a⊂1 over G1a·G2a⊂1.
 | ||
| Fig. 5 Optimized structures of (a) G2a·G2a⊂M1 and (b) G1a·G2a⊂M1Ag at the B3LYP/6-31G(d) and B3LYP/6-31G(d)+LanL2DZ levels. Color scheme: gray (carbon), white (hydrogen), blue (nitrogen), red (oxygen), and pale gray (silver). | ||
Although the homodimer G2a·G2a could not fit within the interior of 1Ag (Fig. 5b), which has a cavity height of 14.5 Å, the heterodimer G1a·G2a was complementary to the interior of 1Ag; this complementarity explains the selective formation of G1a·G2a⊂1Ag.
The regulatable structural features of 1 were studied in solution. AgBF4 was added to a solution of 1 in chloroform-d1, resulting in 1Ag. Then, tetraethylammonium iodide (NEt4I) was added to expel the Ag+ cations from 1Ag as silver iodide (Fig. S18 in the ESI†). This structural interconversion was performed to induce in situ pair exchange between the homo and heterodimeric forms. Fig. 6 shows the change in the signals of the methyl groups of bound G1a and G2a. The mixture of 1 and the guests showed signals corresponding to bound G1a and G2a·G2a (Fig. 6a). When four equivalents of AgBF4 in acetonitrile-d3 were added to the solution, two signals assignable to G1a·G2a⊂1Ag were observed (Fig. 6b). The decomplexation of 1Ag with 8 equivalents of NEt4I resulted in the conversion of G1a·G2a⊂1Ag to (G1a)2⊂1 and G2a·G2a⊂1 (Fig. 6c), although the presence of coexisting acetonitrile-d3 influenced their relative ratio. The original spectrum was recovered by the concentration of the solvents and then adding chloroform-d1 (Fig. 6d). The changes in chemical shifts of the bipyridyl arms (the blue broken lines in Fig. 6a–d) confirmed that the cavity expanded and contracted. A series of experiments showed that bound carboxylic acids underwent pair exchanges in situ by complexation and decomplexation.
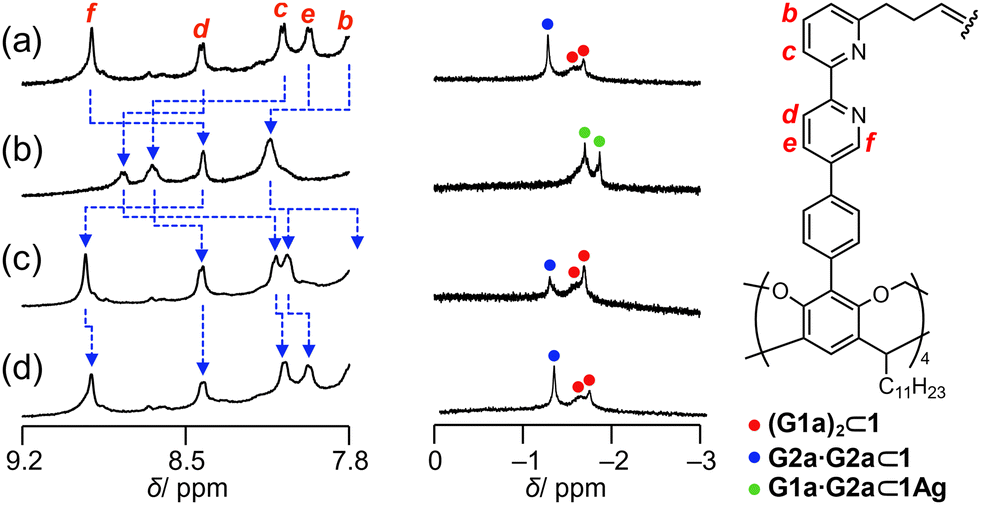 | ||
| Fig. 6 Selected region of the 1H NMR spectra (500 MHz, chloroform-d1, 223 K) of (a) a mixture of G1a (10 mM), G2a (10 mM), and 1 (1.0 mM), (b) after four equivalents of AgBF4 were added in acetonitrile-d3 (60 μL) to the mixture, (c) after 8 equivalents of tetraethylammonium iodide in chloroform-d1 (40 μL) were added to the solution, and (d) after removal of the solvents and dissolution of the resulting solid in chloroform-d1. | ||
In conclusion, resorcinarene-based hemicarcerand 1, which possesses bipyridyl arms, self-sorted the carboxylic acid pairs by contracting and expanding the cavity through the complexation and decomplexation of Ag+ cations in solution. In the expanded form, the homodimeric form of G2a or two molecules of G1a were encapsulated, while the heterodimeric forms of G1a and G2a were selectively organized in 1Ag.
This work was supported by JSPS KAKENHI, Grants-in-Aid for Transformative Research Areas, “Condensed Conjugation” Grant Number JP21H05491 and “Materials Science of Meso-Hierarchy” Grant Number JP23H04873, and Grants-in-Aid for Scientific Research (A) Grant Number JP21H04685. We are also grateful to the KEIRIN JKA, Grant Number 2023M-419. K. H. acknowledges the Grand-in-Aid for the JSPS KAKENHI, Grant number 21J22939.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. F. Perutz, Nature, 1970, 228, 726–734 CrossRef CAS PubMed.
- C. M. Revankar, D. F. Cimino, L. A. Sklar, J. B. Arterburn and E. R. Prossnitz, Science, 2005, 307, 1625–1630 CrossRef CAS PubMed.
- Y.-C. Tang, H.-C. Chang, A. Roeben, D. Wischnewski, N. Wischnewski, M. J. Kerner, F. U. Hartl and M. Hayer-Hartl, Cell, 2006, 125, 903–914 CrossRef CAS PubMed.
- F. U. Hartl, A. Bracher and M. Hayer-Hartl, Nature, 2011, 475, 324–332 CrossRef CAS PubMed.
- H. R. Saibil, W. A. Fenton, D. K. Clare and A. L. Horwich, J. Mol. Biol., 2013, 425, 1476–1487 CrossRef CAS PubMed.
- D. E. Clapham, Cell, 2007, 131, 1047–1058 CrossRef CAS PubMed.
- J. Rebek, Jr., Acc. Chem. Res., 1984, 17, 258–264 CrossRef.
- J.-M. Lehn, Science, 1985, 227, 849–856 CrossRef CAS PubMed.
- S. K. Körner, F. C. Tucci, D. M. Rudkevich, T. Heinz and J. J. Rebek, Chem. – Eur. J., 2000, 6, 187–195 CrossRef.
- J. J. Rebek, Chem. Commun., 2000, 637–643, 10.1039/A910339M.
- S. Shinkai, M. Ikeda, A. Sugasaki and M. Takeuchi, Acc. Chem. Res., 2001, 34, 494–503 CrossRef CAS PubMed.
- L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161–3188 CrossRef CAS PubMed.
- N. C. Gianneschi, M. S. Masar and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825–837 CrossRef CAS PubMed.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed.
- L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261–3277 RSC.
- C. Kremer and A. Lützen, Chem. – Eur. J., 2013, 19, 6162–6196 CrossRef CAS PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- A. M. Lifschitz, M. S. Rosen, C. M. McGuirk and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 7252–7261 CrossRef CAS PubMed.
- A. Jana, S. Bähring, M. Ishida, S. Goeb, D. Canevet, M. Sallé, J. O. Jeppesen and J. L. Sessler, Chem. Soc. Rev., 2018, 47, 5614–5645 RSC.
- J. S. Park and J. L. Sessler, Acc. Chem. Res., 2018, 51, 2400–2410 CrossRef CAS PubMed.
- R. Pinalli, A. Pedrini and E. Dalcanale, Chem. Soc. Rev., 2018, 47, 7006–7026 RSC.
- I. A. Rather, S. A. Wagay, M. S. Hasnain and R. Ali, RSC Adv., 2019, 9, 38309–38344 RSC.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- H.-J. Schneider, Chem. Commun., 2019, 55, 3433–3444 RSC.
- E. Benchimol, B.-N. T. Nguyen, T. K. Ronson and J. R. Nitschke, Chem. Soc. Rev., 2022, 51, 5101–5135 RSC.
- E. L. Piatnitski and K. D. Deshayes, Angew. Chem., Int. Ed., 1998, 37, 970–972 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed.
- F. A. Arroyave and P. Ballester, J. Org. Chem., 2015, 80, 10866–10873 CrossRef CAS PubMed.
- D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS PubMed.
- X. Chi, W. Cen, J. A. Queenan, L. Long, V. M. Lynch, N. M. Khashab and J. L. Sessler, J. Am. Chem. Soc., 2019, 141, 6468–6472 CrossRef CAS PubMed.
- H. Wu, Y. Chen, L. Zhang, O. Anamimoghadam, D. Shen, Z. Liu, K. Cai, C. Pezzato, C. L. Stern, Y. Liu and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 1280–1289 CrossRef CAS PubMed.
- P. N. W. Baxter, R. G. Khoury, J.-M. Lehn, G. Baum and D. Fenske, Chem. – Eur. J., 2000, 6, 4140–4148 CrossRef CAS PubMed.
- S. Hiraoka, K. Harano, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2005, 44, 2727–2731 CrossRef CAS PubMed.
- N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H.-F. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976–12979 CrossRef CAS PubMed.
- D. Ogata and J. Yuasa, Angew. Chem., Int. Ed., 2019, 58, 18424–18428 CrossRef CAS PubMed.
- D. Ajami and J. Rebek, J. Am. Chem. Soc., 2006, 128, 15038–15039 CrossRef CAS PubMed.
- G. Cafeo, F. H. Kohnke, L. Valenti and A. J. P. White, Chem. – Eur. J., 2008, 14, 11593–11600 CrossRef CAS PubMed.
- G. Yu, X. Zhou, Z. Zhang, C. Han, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2012, 134, 19489–19497 CrossRef CAS PubMed.
- K. Kurihara, K. Yazaki, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2017, 56, 11360–11364 CrossRef CAS PubMed.
- J. Mendez-Arroyo, J. Barroso-Flores, A. M. Lifschitz, A. A. Sarjeant, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 10340–10348 CrossRef CAS PubMed.
- J. Mendez-Arroyo, A. I. d’Aquino, A. B. Chinen, Y. D. Manraj and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 1368–1371 CrossRef CAS PubMed.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS PubMed.
- M. Cacciarini, V. A. Azov, P. Seiler, H. Künzer and F. Diederich, Chem. Commun., 2005, 5269–5271, 10.1039/B509990K.
- R. J. Hooley and J. Rebek, Chem. Biol., 2009, 16, 255–264 CrossRef CAS PubMed.
- H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS PubMed.
- M. S. Masar, N. C. Gianneschi, C. G. Oliveri, C. L. Stern, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 10149–10158 CrossRef CAS PubMed.
- H. J. Yoon and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 11590–11591 CrossRef CAS PubMed.
- A. Shivanyuk and J. Rebek, J. Am. Chem. Soc., 2002, 124, 12074–12075 CrossRef CAS PubMed.
- O. Perraud, V. Robert, A. Martinez and J.-P. Dutasta, Chem. – Eur. J., 2011, 17, 4177–4182 CrossRef CAS PubMed.
- N. K. Beyeh, D. P. Weimann, L. Kaufmann, C. A. Schalley and K. Rissanen, Chem. – Eur. J., 2012, 18, 5552–5557 CrossRef CAS PubMed.
- T. Taira, D. Ajami and J. Rebek, Jr., J. Am. Chem. Soc., 2012, 134, 11971–11973 CrossRef CAS PubMed.
- A. Galán, V. Valderrey and P. Ballester, Chem. Sci., 2015, 6, 6325–6333 RSC.
- S. H. A. M. Leenders, R. Becker, T. Kumpulainen, B. de Bruin, T. Sawada, T. Kato, M. Fujita and J. N. H. Reek, Chem. – Eur. J., 2016, 22, 15468–15474 CrossRef CAS PubMed.
- K. Harada, R. Sekiya and T. Haino, Chem. – Eur. J., 2020, 26, 5810–5817 CrossRef CAS PubMed.
- K. Harada, R. Sekiya and T. Haino, J. Org. Chem., 2021, 86, 4440–4447 CrossRef CAS PubMed.
- I. Kojima, M. Yoshida and M. Tanaka, J. Inorg. Nucl. Chem., 1970, 32, 987–995 CrossRef CAS.
- C. Colominas, J. Teixidó, J. Cemelí, F. J. Luque and M. Orozco, J. Phys. Chem. B, 1998, 102, 2269–2276 CrossRef CAS.
- Y. Tsunoda, K. Fukuta, T. Imamura, R. Sekiya, T. Furuyama, N. Kobayashi and T. Haino, Angew. Chem., Int. Ed., 2014, 53, 7243–7247 CrossRef CAS PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00699b |
| This journal is © The Royal Society of Chemistry 2024 |