
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing a σ0π2 singlet ground state carbene from dicationic carbones†
J. Philipp
Wagner

Institut für Organische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076, Tübingen, Germany. E-mail: philipp.wagner@orgchem.uni-tuebingen.de
First published on 28th February 2024
Abstract
Carbenes with a σ0π2 singlet ground state are rare and little is known about their chemistry. Here, we study the potential formation of such carbenes by removal of two electrons from carbones/donor-substituted allenes. The desired electron configuration becomes favorable in the case of bis-diiminium substitution (CAAC motif).
The chemistry of carbenes:CR2, can be understood from the distribution of their two non-bonding electrons over a σ and a π frontier orbital (Fig. 1A).1 When each of these orbitals carries a single electron with parallel spin alignment as in the case of parent methylene (:CH2), a triplet ground state ensues facilitating a radical-like reactivity. Long-lived versions of triplet carbenes can be realized by delocalization of the unpaired electrons and steric protection of the spin centres.2 Likewise, the two nonbonding electrons can pair up in the low-lying σ orbital resulting in a σ2π0 ground state displaying concerted reactivity. This situation gains favour when the σ orbital is lowered by introduction of electronegative substituents while the π orbital is elevated through conjugation with, for instance, lone pairs of electrons. Stable derivatives of such σ2π0 singlet carbenes, especially N-heterocyclic ones, have created a tremendous impact on diverse areas of chemistry.3–7
 | ||
| Fig. 1 The usually high-lying σ0π2 configuration of carbenes (A) becomes the ground state in 2 (B) and 4 (C) facilitating their unusual chemistry. The removal of two electrons from carbones can lead to the potential formation of dicationic carbenes (C). | ||
The σ0π2 configuration is located 59 kcal mol−1 above the ground state in methylene making it much harder to stabilize this state.8 In principle, π-acceptors can be gainfully employed for this task, but this still often leaves the triplet as the ground state like in a recently reported diboryl carbene with a gap of more than 10 kcal mol−1 to the σ0π2 singlet.9 Hoffmann and Borden suggested that the experimentally known 2H-imidazol-2-ylidene can be characterized as a σ0π2 singlet ground state carbene while it was previously rather thought of as a carbodiimide (Fig. 1B).10,11 The uncommon ground state becomes possible because the carbene's lone pair is part of an aromatic sextet and the vacant σ orbital raises in energy through interaction with the in-plane lone pairs of the nitrogen atoms. Our lab has shown that this results in unusual chemistry, such as a preference for least-motion reactions with H2 and alkenes12 as opposed to other ordinary carbenes13,14 as well as a swift reaction with triplet oxygen at cryogenic temperatures15 despite the commonly encountered spin state selectivity of carbenes.16,17
A major breakthrough in the chemistry of σ0π2 carbenes has recently been accomplished with the isolation of rhodium-coordinated diphosphinocarbene 4 building on previous work on ephemeral metalated carbenes (Fig. 1C).18–20 While donation of electron density from the metal centre into the σ orbital stabilizes this carbene, negative hyperconjugation of the π electrons was also discussed to play a role. The importance of the latter interaction suggests that a similar electronic structure could be achieved in carbodiphosphoranes by removal of two electrons to obtain a dicationic carbene (Fig. 1C). Carbodiphosphoranes can be considered as carbones – singlet carbon atoms formally coordinated by two σ-donor ligands bestowing the carbon atom with two lone pairs of electrons.21–24 Carbodicarbenes and carbon suboxide are further experimentally known (‘hidden’) congeners of this compound class25,26 prompting us to explore their viability as carbene precursors through formal removal of one lone pair from the central carbon atom. Therefore, we optimized the dications in Fig. 2 in their respective singlet and triplet states at the B3LYP-D3/def2-TZVPP level of theory and further computed the singlet–triplet gap with the more accurate G4MP2 method where possible.27,28
 | ||
| Fig. 2 Structures of the herein considered singlet dications and their singlet–triplet gaps at the B3LYP-D3/def2-TZVPP and G4MP2 levels of theory. | ||
The simplest dicationic carbone derivative, diphosphonium ion 5, indeed appears to adopt the electronic structure of a carbene displaying carbon-centred frontier orbitals with a doubly occupied σ orbital and vacant π orbital in its singlet state (see ESI† for all orbitals).‡ The C–P bonds are slightly shorter than the expected single bond length29 and the Wiberg bond index (WBI) comes out near unity. The P–C–P angle of 139.7° is rather large for a singlet carbene which becomes understandable from the repelling substituents of like charge. The wide angle favours the triplet making it the ground state by a substantial 23.1 kcal mol−1 (G4MP2). Since the lowest singlet corresponds to the σ2π0 configuration, we performed a state averaged NEVPT2 calculation with a CAS(2,2) reference to study the σ0π2 state, locating it at 73.8 kcal mol−1 above the ground state. This result reveals that negative hyperconjugation to phosphonium groups is far from sufficient to stabilize the σ0π2 state and re-emphasizes the role of the metal in 4.18 A comparable geometric and electronic structure is observed for the methylated diphosphonium ion 6.
We reasoned that a more powerful electron acceptor is required, turning to carbon suboxide dication 7 in the following. The bent singlet state again assumes a σ2π0 configuration with a large singlet–triplet gap of 22.3 kcal mol−1. Natural resonance theory (NRT) predicts a dominant carbene structure with a weight of 63.6% in which two acylium ions carry the positive charges. The bending of the molecule is in line with the presence of a lone pair at the central carbon atom. The molecule is stabilized by in-plane delocalization of the carbene's nonbonding electrons producing mesomeric structures with a ketene unit. This might explain the Wiberg bond index of 1.328 and the rather short C–C bond distance of 1.38 Å, which is closer in length to a typical double than a single bond.
The dicationic carbodicarbene 8 displays a much smaller singlet triplet gap of 3.5 kcal mol−1. The outermost, carbon-centred orbitals are of σ- and π-type with the former being doubly occupied. Although the NRT analysis predicts a rather complex set of structures, drawing a carbene appears reasonable since the imidazolium substituents can well support the positive charges.
The short C–C bond distances of 1.37 Å and the Wiberg bond index well above one cast doubt on the assignment of a carbene structure featuring two single bonds to the central atom in the case of 8. Close inspection of the molecular orbitals reveals the presence of a doubly occupied π-type orbital that is all-bonding along the central C–C–C unit and not counteracted by a higher-lying filled orbital of strongly antibonding character. While the structure of the bis-benzimidazolium carbene 9 is comparable to 8, dication 10 with its saturated backbone should not be regarded as a carbene. The latter molecule's LUMO still corresponds to a carbon-centred π orbital, but the two highest occupied orbitals are associated with the nitrogen atoms’ lone pairs; the carbene σ orbital only represents the HOMO−2. In addition, the central C–C–C unit of dication 10 does not linearize in its triplet state as seen for molecules 5–9.
So far, some of the considered carbone dications can well be considered as carbenes, but their singlet states pertain to the conventional σ2π0 configuration. Hence, we reckoned that the carbone's substituents (ligand L) are required to be stronger π-acceptors which is seen realized in the cyclic alkyl amino carbene (CAAC) motif.30 Although the neutral precursor might not necessarily correspond to a carbone but rather to a donor-substituted, linear allene, the singlet dication 11 indeed displays the desired σ0π2 configuration (Fig. 3, top). The π orbital presents a significant coefficient of the atomic 2p orbital at the carbene carbon atom and indicates resonance with the neighbouring iminium ion groups acting as powerful electron sinks to stabilize the unconventional singlet state. Natural resonance theory confirms this analysis assigning the carbene as the dominant mesomeric structure (weight 30.2%) with delocalization of the lone pair facilitated by the iminium ions.
 | ||
| Fig. 3 Frontier orbitals of 11 and its dominant resonance structures. | ||
Despite this initial success, carbene 11 still prefers a triplet ground state by 10.9 kcal mol−1. This motivated us to employ a second mode of stabilization targeting the carbene's σ orbital. A look at the LUMO in Fig. 3 shows that the vacant orbital is perfectly aligned for hyperconjugation with the five-membered rings’ vicinal C–C bonds. Hence, we decided to bring the silicon β effect to use and placed SiMe2 groups at the relevant position yielding dication 12. This additional mode of stabilization helped to tip the balance finally predicting a σ0π2 singlet ground state for 12. The singlet–triplet gap amounts to −5.7 kcal mol−1 at the G4MP2 level of theory.
Natural resonance theory produces a number of structures featuring silylium ion centres which is understandable from the elongated carbon–silicon bonds in 12 (2.06 Å). The central C–C bonds assume the length of a typical double bond bringing the carbene character of the dication once again into question. Therefore, we decided to verify the nature of the molecule by studying its single bond insertion reactivity. The relevant transition state for the insertion of the central carbon atom into the dihydrogen molecule is depicted in Fig. 4 and comes with a barrier of 21.9 kcal mol−1. As expected for σ0π2 carbenes,12 the reaction mode corresponds to a σ-approach but deviates from the least-motion trajectory because the reaction centre is blocked by the two N-methyl substituents. The favourable bond insertion reactivity corroborates the nature of 12 as a σ0π2 singlet ground state carbene.
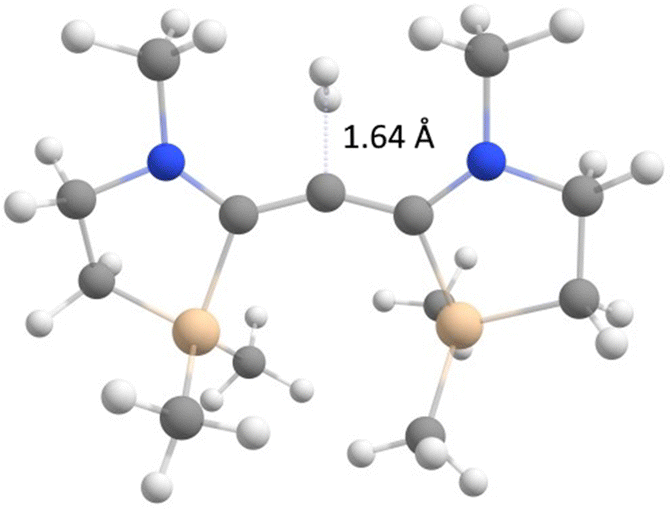 | ||
| Fig. 4 Transition state of the hydrogen insertion reaction of 12. | ||
In summary, we have demonstrated that some carbone dications can be viewed as carbenes. The unconventional σ0π2 state becomes energetically accessible by stabilizing the π orbital through conjugation with iminium ions (CAAC scaffold) and destabilizing the σ orbital via hyperconjugation with carbon–silicon bonds. The experimental realization of such carbenes appears to be a worthwhile but challenging endeavour.
This work was generously funded by the Fonds der Chemischen Industrie in the form of a Liebig fellowship. Computations were performed on the BwForCluster JUSTUS 2 facilitated by bwHPC and the DFG (INST 40/575-1 FUGG). The author thanks Prof. Holger Bettinger for his valuable support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. B. Eisenthal, R. A. Moss and N. J. Turro, Science, 1984, 225, 1439–1445 CrossRef CAS.
- H. Tomioka, E. Iwamoto, H. Itakura and K. Hirai, Nature, 2001, 412, 626–628 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1989, 28, 621–622 CrossRef.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS.
- D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS.
- A. Kalemos, T. H. DunningJr., A. Mavridis and J. F. Harrison, Can. J. Chem., 2004, 82, 684–693 CrossRef CAS.
- Y. Shibutani, S. Kusumoto and K. Nozaki, J. Am. Chem. Soc., 2023, 145, 16186–16192 CrossRef CAS.
- G. Maier and J. Endres, Chem. – Eur. J., 1999, 5, 1590–1597 CrossRef CAS.
- B. Chen, A. Y. Rogachev, D. A. Hrovat, R. Hoffmann and W. T. Borden, J. Am. Chem. Soc., 2013, 135, 13954–13964 CrossRef CAS.
- S. F. Clewing and J. P. Wagner, J. Org. Chem., 2021, 86, 15247–15252 CrossRef CAS.
- R. Hoffmann, J. Am. Chem. Soc., 1968, 90, 1475–1485 CrossRef CAS.
- A. E. Keating, S. R. Merrigan, D. A. Singleton and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 3933–3938 CrossRef CAS.
- J. P. Wagner, J. Am. Chem. Soc., 2022, 144, 5937–5944 CrossRef CAS.
- W. Sander, Angew. Chem., Int. Ed. Engl., 1990, 29, 344–354 CrossRef.
- J. P. Wagner, Phys. Chem. Chem. Phys., 2022, 24, 25834–25841 RSC.
- C. Hu, X.-F. Wang, J. Li, X.-Y. Chang and L. L. Liu, Science, 2024, 383, 81–85 CrossRef CAS PubMed.
- J. Vignolle, H. Gornitzka, L. Maron, W. W. Schoeller, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 978–985 CrossRef CAS.
- J. Ruiz, M. E. G. Mosquera, G. García, E. Patrón, V. Riera, S. García-Granda and F. Van der Maelen, Angew. Chem., Int. Ed., 2003, 42, 4767–4771 CrossRef CAS.
- F. Ramirez, N. B. Desai, B. Hansen and N. McKelvie, J. Am. Chem. Soc., 1961, 83, 3539–3540 CrossRef CAS.
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS.
- G. Frenking, R. Tonner, S. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, Chem. Soc. Rev., 2014, 43, 5106–5139 RSC.
- G. Frenking, M. Hermann, D. M. Andrada and N. Holzmann, Chem. Soc. Rev., 2016, 45, 1129–1144 RSC.
- R. Tonner and G. Frenking, Angew. Chem., Int. Ed., 2007, 46, 8695–8698 CrossRef CAS.
- C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206–3209 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Chem. Phys., 1994, 98, 11623–11627 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 127, 124105 CrossRef.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Optimized geometries and energies. See DOI: https://doi.org/10.1039/d4cc00492b |
| ‡ We note that the singlet dications considered herein do not display a plane of symmetry with the exception of 7 rendering a rigorous definition of σ and π orbitals unattainable. |
| This journal is © The Royal Society of Chemistry 2024 |