
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of sydnonimines from sydnones and their use for bioorthogonal release of isocyanates in cells†
Judith
Baudet
a,
Emilie
Lesur
a,
Maxime
Ribéraud
a,
Arnaud
Chevalier
 b,
Timothée
D’Anfray
a,
Pierre
Thuéry
c,
Davide
Audisio
a and
Frédéric
Taran
*a
b,
Timothée
D’Anfray
a,
Pierre
Thuéry
c,
Davide
Audisio
a and
Frédéric
Taran
*a
aDépartement Médicaments et Technologies pour la Santé (DMTS), CEA, INRAE, SCBM, Université Paris Saclay, Gif-sur-Yvette 91191, France. E-mail: frederic.taran@cea.fr
bInstitut de Chimie des Substances Naturelles, CNRS, UPR 2301, Université Paris-Saclay, 91198, Gif-sur-Yvette, France
cCEA, CNRS, NIMBE, Université Paris-Saclay, 91191, Gif-sur-Yvette, France
First published on 29th February 2024
Abstract
In this article, we report the synthesis of sydnonimines from sydnones and their use as dipoles for fast click-and-release reactions. The process relies on nucleophilic aromatic substitution of aliphatic and aromatic amines with triflated sydnones. This new methodology allowed the preparation of functionalised sydnonimine probes that are otherwise difficult to prepare. These probes were then used to release a drug and a fluorescent aromatic isocyanate inside living cells.
Sydnonimines (SIs) belong to the mesoionic family, closely related to sydnones with the key distinction of containing a nitrogen atom at position 6. Discovered in the 1950s1 and further developed in the 1970s for their biological properties, some of them have been approved as drugs.2 Renewed interest in SIs has recently emerged due to their ability to undergo chemoselective cycloaddition reactions with strained alkynes, termed SPSIC (for Strained Promoted SydnonImine Cyclooctyne Cycloaddition).3 This reaction mechanism involves a two-step process: a [3+2] cycloaddition followed by a retro-Diels–Alder, yielding a pyrazole clicked product and an isocyanate released product. SPSIC exhibits promise as a novel bioorthogonal click-and-release tool applicable in target fishing,4 bioconjugation,5 cell imaging6 and drug release in complex biological media.7 The speed of the reaction is remarkably affected by the substituents at position 6 of the SI core: the rate constant of the SPSIC reaction can be enhanced by 6 orders of magnitude simply by replacing electron-withdrawing groups by electron donating groups at this position (Scheme 1).8 Our group has recently reported on novel SIs bearing alkyl or aryl groups at position 6 exhibiting remarkable kinetics for the SPSIC reaction (k up to 1000 M−1 s−1).8 The synthesis of these compounds involves either reductive amination or cyclodehydration of nitrosoamides as key steps (Scheme 1). To expand the scope of synthetically accessible SIs, we aimed to develop a method enabling direct modification at position 6 via nucleophilic aromatic substitution (SNAr) of sydnones with amines, which are accessible both synthetically and commercially. Such a method would enable SIs to be obtained in a single step with the possibility of significant structural diversity.
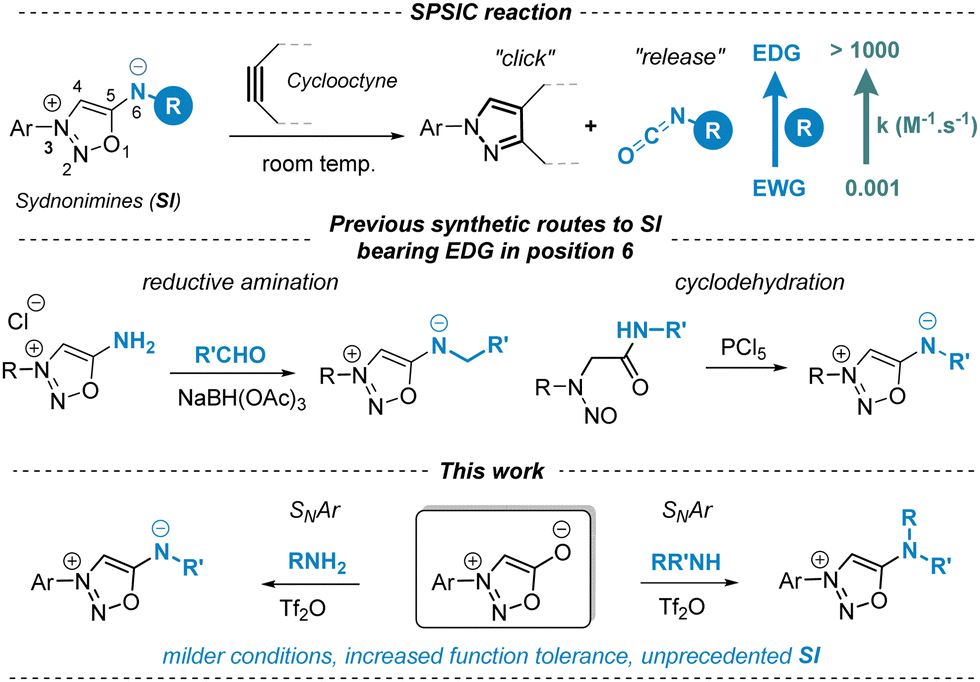 | ||
| Scheme 1 SPSIC reaction and synthetic routes to 6-N-alkyl and 6-N-aryl-SI. | ||
The oxygen at position 6 of sydnones is known for its poor nucleophilicity and only very strong electrophiles are able to react with it.9 Nonetheless, aromatic nucleophilic substitutions at this position were reported in 1985 after the activation of this oxygen atom by Tf2O and the addition of a malononitrile carbanion.10 This reaction was recently extended to other activated methylene nucleophiles by the group of A. Schmidt to synthesise and study a series of sydnone methide compounds.11 However, to date no implementation of this methodology has been reported for the introduction of alternative nucleophiles. Inspired by this work, we hypothesised that amines could serve as nucleophiles in this reaction, offering a new route to SIs (Scheme 1). According to literature data,10 the reaction of sydnones with Tf2O yields a mixture of sydnone 5-triflates I and bis-sydnone ethers II, found to be very sensitive towards minute traces of water. Both species are in principle able to react with nucleophiles. Prior to investigating the reaction with amines, we initiated a study to elucidate this activation step through quenching experiments with isotopically labelled H218O (Scheme 2).
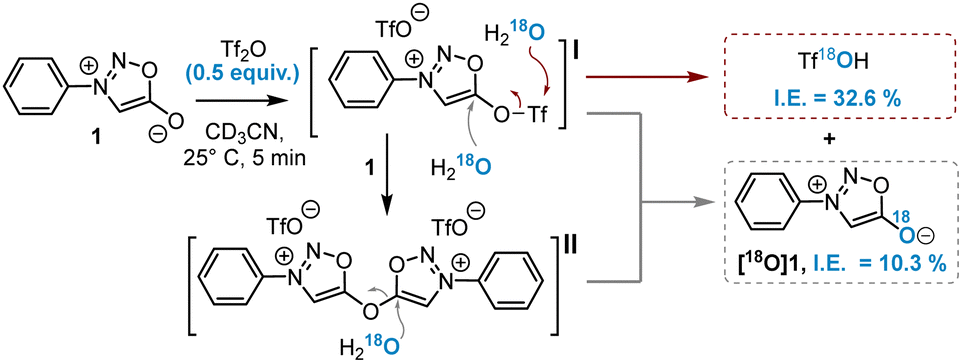 | ||
| Scheme 2 Reaction of sydnones with Tf2O. I.E.: isotopic enrichment. | ||
The activation of sydnone 1 was carried out using 0.5 equiv. of Tf2O to favour the formation of intermediate II. As a consequence, after quenching with H218O a maximum of 50% isotopic enrichment (I.E.) of labelled sydnone [18O]1 or Tf18OH could be expected. 19F-NMR monitoring showed complete consumption of Tf2O before addition of H218O, while 1H-NMR indicated the formation of two new sydnone species (Fig. S1, ESI†). 19F-NMR and HRMS analysis just after the addition of H218O showed Tf18OH as the major labelled product, which can be generated only from the hydrolysis of I (Fig. S2, ESI†). 18O-labelled sydnone [18O]1, formed by SNAr reaction either on I or II, represented only a quarter of the total labeled compounds. Control experiments confirmed that no 18O exchange occurred either on sydnone or on the triflate salt under the reaction conditions (Fig. S3, ESI†). These results proved that I is the main active species formed during the reaction and, as expected, that the SNAr reaction on I competes with the nucleophilic attack on the sulphur atom of the triflate moiety. This side reaction could pose a significant challenge when amine nucleophiles are used on I.
From these findings, we explored the reaction with aliphatic and aromatic amines using benzylamine and aniline as model substrates (Table 1). The SNAr reactions of both model amines on triflate I were found to be very fast: completion of the reactions was observed after 10 min even at room temperature showing the high reactivity of triflate I. Gentle heating at 40 °C allowed a significant increase of the yield, but higher temperatures were detrimental to the reaction due to the degradation of the substrates. The presence of a base is beneficial to the reaction, TEA being the most effective. Finally, the influence of the solvent was investigated revealing CH3CN as the most suitable. Despite these efforts, the yields plateaued around 50% due to the side reaction generating triflated amines 2 and to purification issues. Attempts to use alternative activation methods for sydnones besides Tf2O (Ts2O, Ns2O…) to minimize the formation of 2 were unsuccessful.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | RNH2 | Base | Temp. (°C) | SI (yield)a | 2 (yield)b |
| a Crude yields determined by NMR for entries 1–11 and isolated yields for entries 12–16. n.d. = not detected. b Crude yields determined by UPLC-MS. | ||||||
| 1 | MeCN | BnNH2 | — | 25 | 30% | 40% |
| 2 | MeCN | BnNH2 | DiPEA | 25 | 31% | 24% |
| 3 | MeCN | BnNH2 | DiPEA | 40 | 39% | 39% |
| 4 | MeCN | BnNH2 | DiPEA | 60 | n.d. | n.d. |
| 5 | MeCN | BnNH2 | DABCO | 40 | 47% | 3% |
| 6 | MeCN | BnNH2 | Pyridine | 40 | n.d. | n.d. |
| 7 | MeCN | BnNH 2 | TEA | 40 | 50% | 36% |
| 8 | DMF | BnNH2 | TEA | 40 | n.d. | n.d. |
| 9 | THF | BnNH2 | TEA | 40C | 45% | n.d. |
| 10 | CHCl3 | BnNH2 | TEA | 40 | 42% | 11% |
| 11 | Toluene | BnNH2 | TEA | 40 | 50% | n.d. |
| 12 | MeCN | PhNH 2 | TEA | 40 | 44% | 19% |
| 13 | MeCN | PhNH2 | TEA | 60 | 41% | 25% |
| 14 | MeCN | PhNH2 | TEA | 80 | 40% | 17% |
| 15 | THF | PhNH2 | NaH | 40 | n.d. | n.d. |
| 16 | THF | PhNH2 | tBuOK | 40 | n.d. | n.d. |
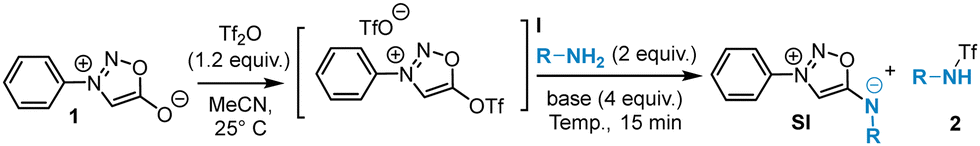
Despite the challenges, we were intrigued by the potential synthetic utility of this reaction and explored its scope further (Scheme 3).
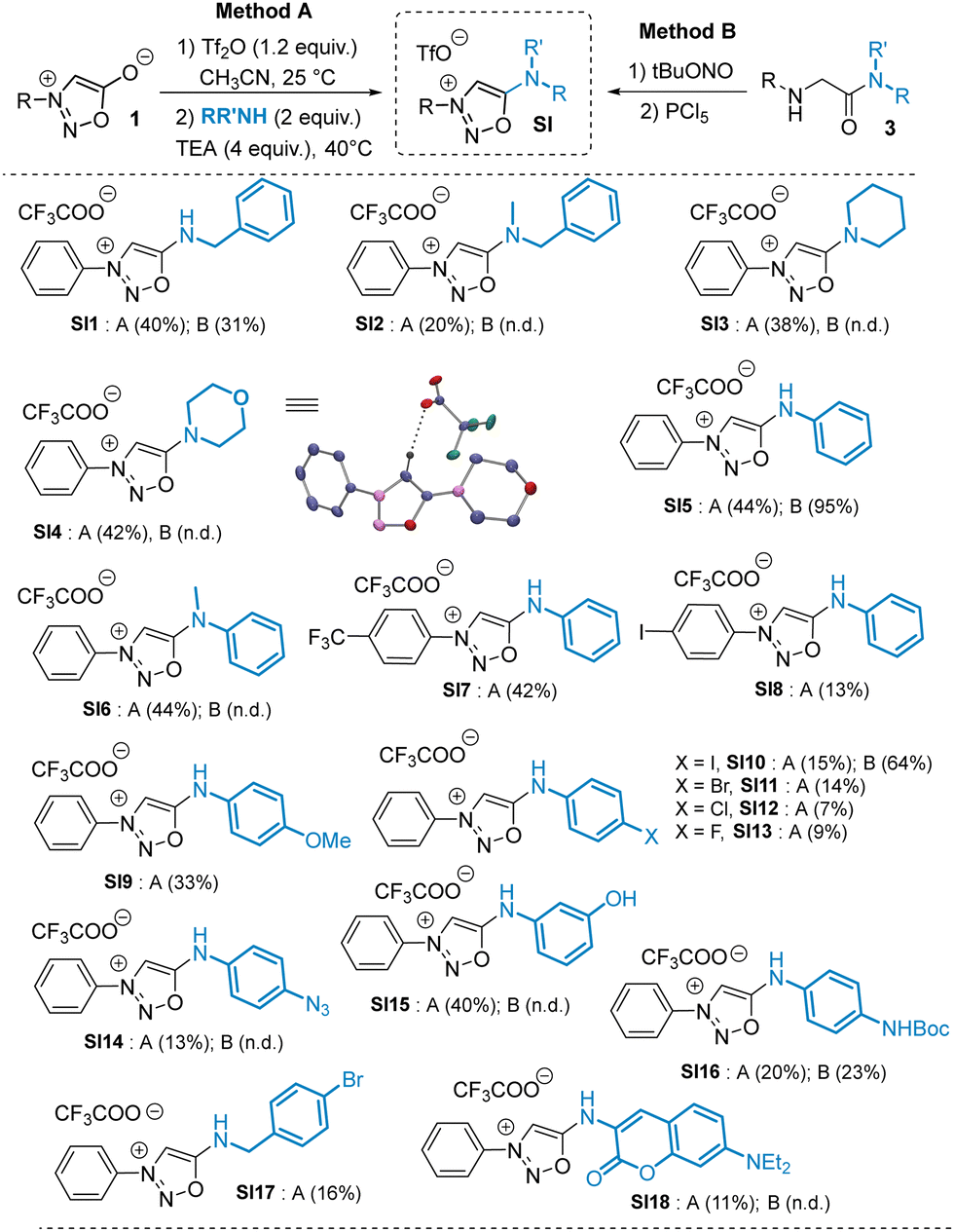 | ||
| Scheme 3 Scope of the reaction. Reaction conditions: (1) TF2O 1.2 equiv., MeCN – 25 °C, (2) RR′NH 2.0 equiv. TEA 4 equiv. MeCN – 40 °C. n.d. = not detected. The trifluoroacetate anion comes from the reversed phase purification. | ||
Smooth SNAr reactions were observed with both aliphatic and aromatic amines bearing electron rich or electron-poor substituents. To compare the SNAr reaction (method A) with our previously reported cyclodehydration approach (method B),8 we synthesized a series of SIs using both methods. The SNAr procedure proved to be a valuable complement to method B and allowed the synthesis of SIs otherwise difficult or impossible to prepare. Although the SNAr approach gave low to moderate yields, it tolerates a variety of functional groups such as phenol, azide and carbamate, which are incompatible with method B. Interestingly, secondary amines are also good substrates for the SNAr reaction, yielding unprecedented compounds bearing an exocyclic tertiary amine at position 6 of the sydnonimine core. These derivatives, inaccessible via method B, are not mesoionics and thus represent novel chemical species for exploration in cycloaddition reactions with strained alkynes. We thus conducted kinetic experiments with model substrates SI5 and SI6, which differ only in the presence of a methyl group, in the presence of the cyclooctyne DBCO (Scheme 4).
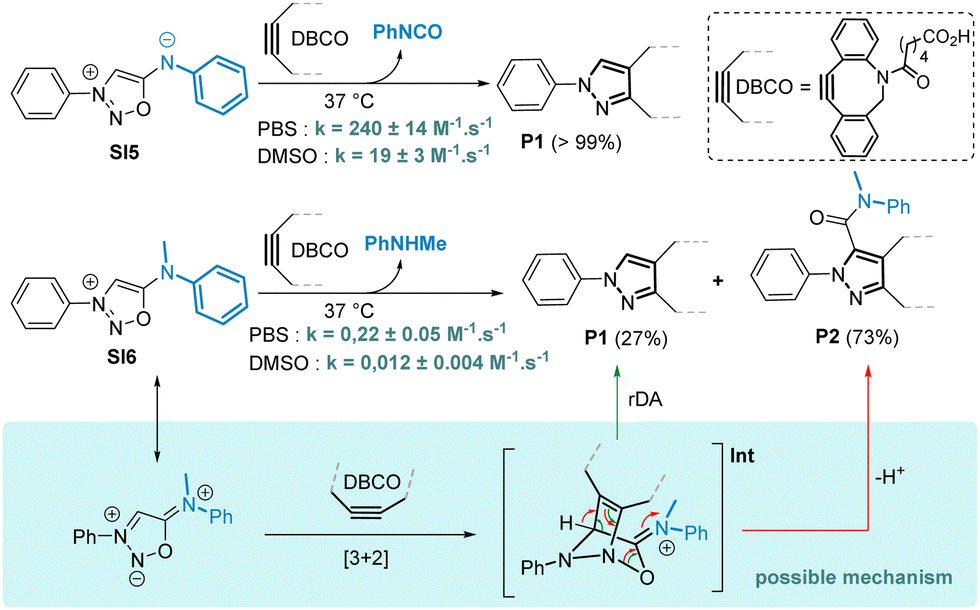 | ||
| Scheme 4 Reaction of sydnonimines SI5 and SI6 with DBCO. Proposed mechanism for the formation of P1 and P2 from SI6. The rate constants were determined by UV monitoring, [SI5] = [SI6] = 100 μM, DBCO 1.0 equiv. in 0.1 M PBS pH = 7.4 or in DMSO. Yields of pyrazoles were estimated by 1H NMR monitoring in DMSO-d6. | ||
As anticipated, the kinetic behaviour of the two compounds exhibited drastic differences. Compound SI6 reacted 923 times slower in reaction with DBCO in PBS compared to SI5. In addition, the reaction of DBCO with SI6 produced not just one but two pyrazole products P1 and P2. Both pyrazoles were formed at the same speed with a kinetic constant of k = 0.22 ± 0.05 M−1 s−1 in PBS, suggesting that the mechanism involves a common intermediate whose formation is the rate determining step. We postulate that the formation of intermediate Int resulting from a [3+2] cycloaddition on SI6 can undergo a rDA reaction to form pyrazole P1. On the other hand, Int can also evolve after proton abstraction to generate pyrazole P2 (Scheme 4). As the formation of pyrazole P1 is concomitant with the release of the secondary amine attached in position 6 of the SI core, a possible application of this chemistry may lie in the release of drugs attached to this position. To evaluate this hypothesis, we conducted proof-of-concept experiments. SI19, obtained in 20% yield using the SNAr reaction, was designed to release the antidepressant drug Paroxetin upon reaction with DBCO (Scheme 5). The SPSIC reaction was conducted in aqueous conditions at pH 7.4, 25 °C and monitored by UPLC. The results confirmed a significant rate constant of k = 1.12 ± 0.21 M−1 s−1 but incomplete release of the drug over time (∼13% release, Fig. S13, ESI†). This lack of release efficiency may be attributed to the formation of Int2 that cannot undergo the rDA-promoted release of the Paroxetin. To avoid this, we then synthesised SI20 and SI21 substituted by a Cl atom and a methyl group, respectively, at position 4 of the SI core in order to prevent the proton abstraction step. Although SI21 proved to be unreactive, SI20 allowed slow but efficient drug release upon reaction with DBCO (Scheme 5 and Fig. S16, ESI†). Although improvements are clearly required, these SI-protected drugs might be interesting in future bioorthogonal decaging strategies.
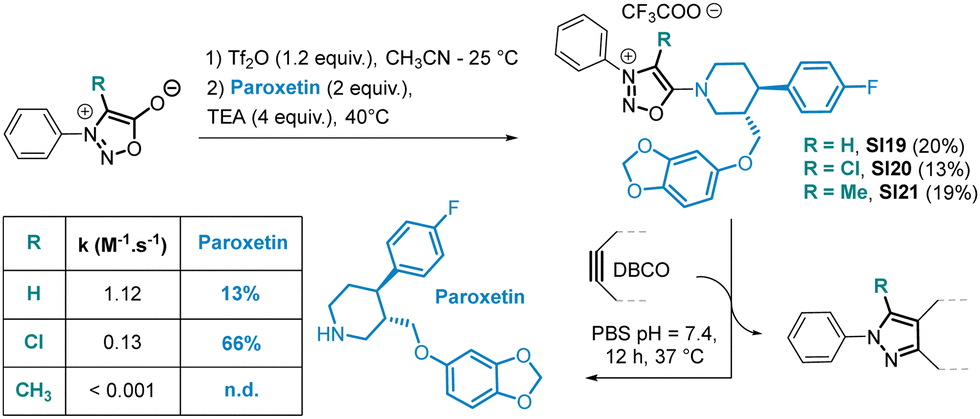 | ||
| Scheme 5 Release of paroxetin from SI19–21; monitoring was carried out by UPLC, 4-nitro-2-fluorophenol as an internal standard. Reaction conditions: [SI] = 100 μM, DBCO 1.0 equiv. in PBS pH = 7.4, 25 °C. | ||
We then focused our interest on the coumarin-containing compound SI18, which should generate a fluorescent isocyanate upon SPSIC reaction. Interestingly, we found that SI18 reacted as a fluorogenic probe with DBCO to form an isocyanate, the fluorescence of which is slightly higher than the starting sydnonimine (turn-on around × 5, Fig. 1).
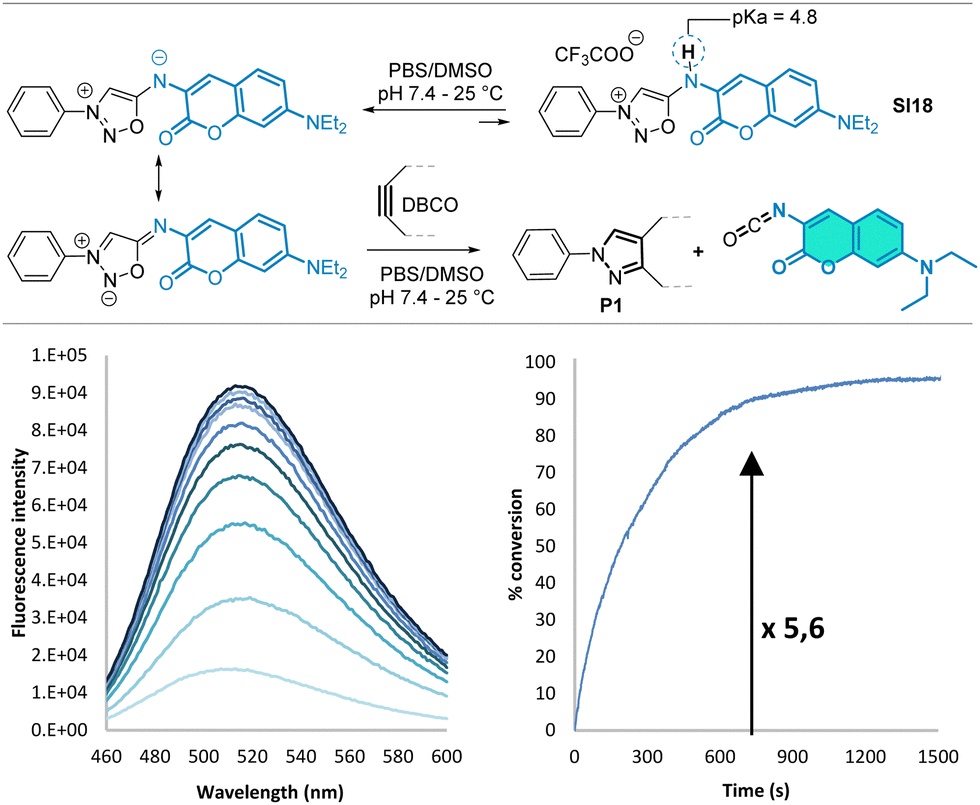 | ||
| Fig. 1 SPSIC reaction of probe SI18 with DBCO under physiological conditions. Kinetics were monitored by fluorescence, λex = 404 nm, λem = 510 nm. Reactions conditions: 37 °C, [SI18] = 10 μM, DBCO 1.5 equiv. in PBS pH = 7.4. | ||
The pKa of compound SI18 was determined at 4.8 ± 0.1 (Fig. S8 and S10, ESI†). Accordingly, SI18 is mainly deprotonated at physiological pH generating a highly reactive mesoionic species. This was confirmed by fluorescence monitoring of the SPSIC reaction of SI18 with DBCO: the kinetic constant was found to be k = 533 ± 27 M−1 s−1 meaning complete click-and-release reaction within minutes when the compounds are used in μM concentrations (Fig. S12, ESI†). We then studied whether this probe may be used to release fluorescent isocyanates inside living cells (Fig. 2A). A549 cells were incubated for 2 h with SI18. After washing, SPSIC reaction was initiated by the addition of DBCO (250 μM). Careful monitoring using wide field microscopy (Fig. S17 and S18, ESI†) over 2 h showed a 4-fold increase in fluorescence signal intensity achieved after 30 min reaction time (Fig. 2B). The confocal microscopy images shown in Fig. 2D indicate a significant increase in fluorescence intensity after addition of DBCO. Interestingly, this labelling was resistant to several successive washing steps (Fig. 2C and Fig. S19, ESI†), suggesting irreversible fluorescent labelling of the cells. In the meantime, the same washing process induced a significant drop of the fluorescence signal intensity in the case of untreated SI18. Altogether, these results confirmed the effective biorthogonal release of the coumarin-isocyanate derivative inside the cells leading to persistent labeling of the cells.
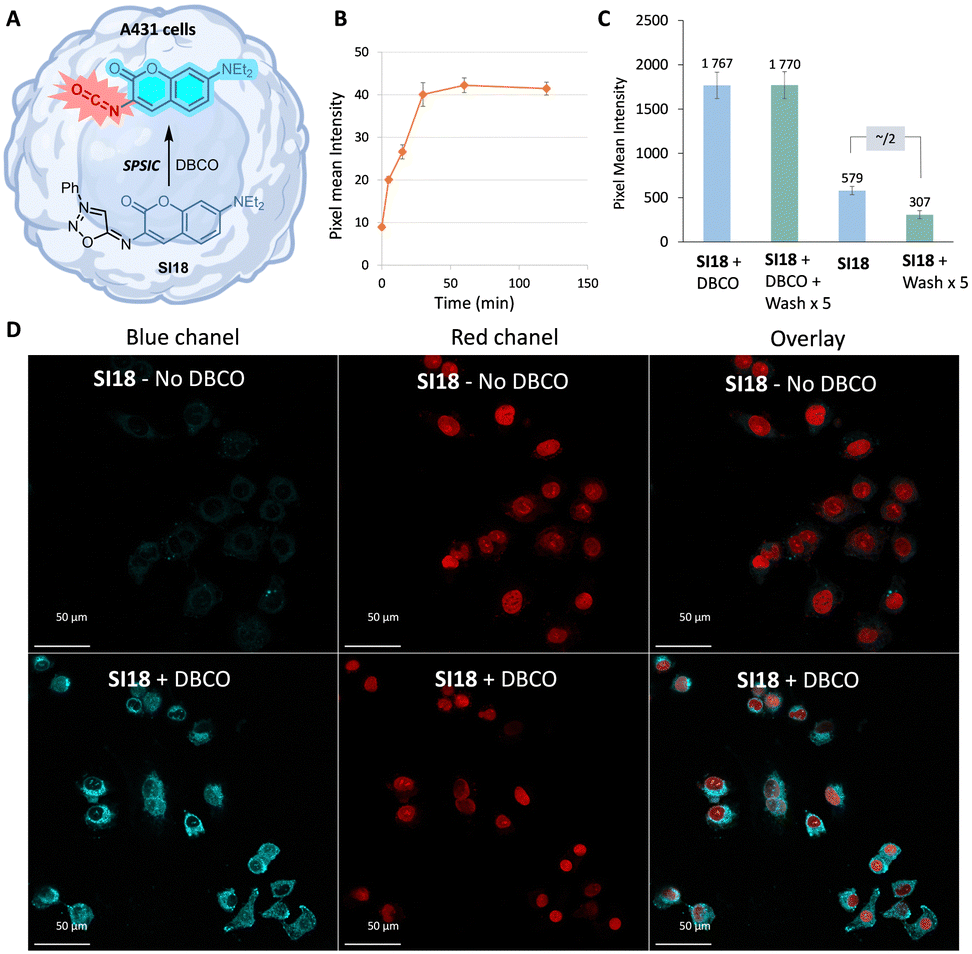 | ||
| Fig. 2 SPSIC reaction inside living A549 cells. Cells were washed before the addition of DBCO. (A) Schematic representation of isocyanate release. (B) Fluorescence intensity evolution with time in A549 cells after the addition of DBCO. (C) Fluorescence intensity after 2 h in the presence or absence of DBCO and with or without cell washing. (D) Confocal microscopy images of cells treated with probe SI18 in the presence or absence of DBCO. Conditions: [SI18] = 50 μM, incubation for 2 h then wash with PBS, addition of [DBCO] = 20 μM during 1 h. | ||
In summary, we have developed a new method for synthesizing sydnonimines that were previously difficult to obtain. Although the yields need improvement, this method's simplicity and tolerance to functional groups make it advantageous compared to existing methods. We have successfully synthesized unique SIs with tertiary amines at position 6, tested them in cycloaddition reactions with DBCO, and explored potential applications in drug release. Additionally, the SNAr reaction allowed access to a fluorogenic probe for efficient fluorescence labeling inside living cells. Such compounds allow the release of fluorescent isocyanates in cells, which can be exploited to visualize bionucleophiles and to permanently label living cells holding promise for advancing our understanding of cellular processes and protein dynamics.
This work was supported by the Agence Nationale de la Recherche (ANR-23-CE07-0041-01). The project benefited from the Imagerie-Gif core facility supported by l’Agence Nationale de la Recherche (ANR-11-EQPX-0029/Morphoscope, ANR-10-INBS-04/FranceBioImaging; ANR-11-IDEX-0003-02/Saclay Plant Sciences). The authors thank David-Alexandre Buisson, Sabrina Lebrequier and Amélie Goudet for excellent analytical support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Brookes and J. Walker, J. Chem. Soc., 1957, 4409–4416 RSC.
- E. Y. Khmel’nitskaya, V. I. Levina, L. A. Trukhacheva, N. B. Grigoriev, V. N. Kalinin, I. A. Cherepanov, S. N. Lebedev and V. G. Granika, Russ. Chem. Bull., 2004, 53, 2840–2844 CrossRef.
- S. Bernard, D. Audisio, M. Riomet, S. Bregant, A. Sallustrau, L. Plougastel, E. Decuypere, S. Gabillet, R. A. Kumar, J. Elyian, M. N. Trinh, O. Koniev, A. Wagner, S. Kolodych and F. Taran, Angew. Chem., Int. Ed., 2017, 56, 15612–15616 CrossRef CAS PubMed.
- S. Saidjalolov, E. Braud, Z. Edoo, L. Iannazzo, F. Rusconi, M. Riomet, A. Sallustrau, F. Taran, M. Arthur, M. Fonvielle and M. Etheve-Quelquejeu, Chem. – Eur. J., 2021, 27, 7687–7695 CrossRef CAS PubMed.
- I. Dovgan, A. Hentz, O. Koniev, A. Ehkirch, S. Hessmann, S. Ursuegui, S. Delacroix, M. Riomet, F. Taran, S. Cianférani, S. Kolodych and A. Wagner, Chem. Sci., 2020, 11, 1210–1215 RSC.
- M. Riomet, K. Porte, A. Wijkhuisen, D. Audisio and F. Taran, Chem. Commun., 2020, 56, 7183–7186 RSC.
- (a) Z. Shao, W. Liu, H. Tao, F. Liu, R. Zeng, P. A. Champagne, Y. Cao, K. N. Houk and Y. Liang, Chem. Commun., 2018, 54, 14089–14092 RSC; (b) M. Feng, L. Madegard, M. Riomet, M. Louis, P. A. Champagne, G. Pieters, D. Audisio and F. Taran, Chem. Commun., 2022, 58, 8500–8503 RSC.
- M. Ribéraud, K. Porte, A. Chevalier, L. Madegard, A. Rachet, A. Delaunay-Moisan, F. Vinchon, P. Thuéry, G. Chiappetta, P. A. Champagne, G. Pieters, D. Audisio and F. Taran, J. Am. Chem. Soc., 2023, 145(4), 2219–2229 CrossRef PubMed.
- S. Wiechmann, T. Freese, M. H. H. Drafz, E. G. Hübner, J. C. Namyslo, M. Niegerb and A. Schmidt, Chem. Commun., 2014, 50, 11822–11824 RSC.
- S. Araki, J. Mizuya and Y. Butsugan, J. Chem. Soc., Perkin Trans. 1, 1985, 2439–2441 RSC.
- S. Mummel, F. Lederle, E. G. Hübner, J. C. Namyslo, M. Nieger and A. Schmidt, Angew. Chem., Int. Ed., 2021, 60, 18882–18887 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2329777. See DOI: https://doi.org/10.1039/d4cc00490f |
| This journal is © The Royal Society of Chemistry 2024 |