
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric copper-catalyzed alkynylallylic monofluoroalkylations with fluorinated malonates†
Han-Yu
Lu
abc,
Zi-Han
Li
ac,
Guo-Qiang
Lin
ab and
Zhi-Tao
He
 *cde
*cde
aState Key Laboratory of Chemical Biology, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai, 200032, China
bSchool of Physical Science and Technology, ShanghaiTech University, Shanghai, 201210, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai, 200032, China. E-mail: hezt@sioc.ac.cn
dSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China
eNingbo Zhongke Creation Center of New Materials, Ningbo, 315899, China
First published on 18th March 2024
Abstract
The unprecedented copper-catalyzed asymmetric alkynylallylic monofluoroalkylation reaction is described via the use of 1,3-enynes and fluorinated malonates. A series of 1,4-enynes bearing a monofluoroalkyl unit are achieved in high yields, excellent regio- and enantioselectivity and high E/Z selectivity. The asymmetric propargylic monofluoroalkylation is also developed. The reliability and synthetic value of the work are highlighted by a gram-scale test and a couple of downstream transformations. Preliminary mechanistic studies unveil a negative nonlinear effect for the catalytic process.
Copper-catalyzed asymmetric propargylic substitution reactions have emerged as a reliable and valuable route to construct stereogenic centers.1 A series of nucleophiles have been efficiently introduced to the propargylic position to construct C–C, C–N, C–S, and C–O bonds with high stereocontrol (Scheme 1(a), left).2 As the F-containing motif has been widely used in the design of biologically active molecules,3 the construction of stereocenters bearing fluorine atoms via Cu-catalyzed propargylation represents a novel route to achieve such optically active skeletons but related studies are very limited. Zhang et al. sequentially developed efficient catalytic systems for the preparation of propargylic CF3, RCF2 and SCF3 units.4–6 In addition, Tang recently described an elegant protocol to introduce OCF3-based stereocenters via propargylic substitution.7 However, the propargylic monofluoroalkylation and related processes remain unexplored (Scheme 1(a), right).
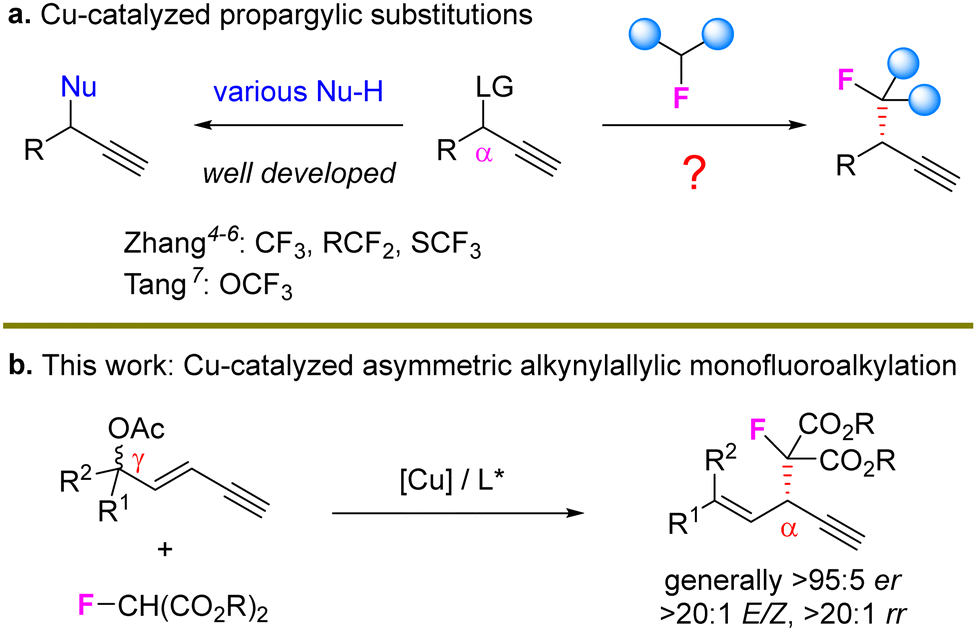 | ||
| Scheme 1 Cu-catalyzed propargylic substitutions. | ||
Different from the typical propargylic substitution requiring an α-leaving group to guarantee the formation of a critical Cu-allenylidene intermediate, an alkyne bearing a remote leaving group is usually not considered as a suitable substrate for propargylation. In 2022, Fang's group first reported a regiodivergent but non-asymmetric Cu-catalyzed alkynylallylic substitution model by using 1,3-enyne with a γ-leaving group as the substrate.8 Then, we described a highly enantioselective process.9 In our work, various enantioenriched 1,4-enyne10 skeletons were achieved in high yields and enantiocontrol. Later, Xu and Qi developed an elegant in situ substitution route to prepare various spirocycles in high enantioselectivity.11 After these initial works, we further established another type of remote stereocontrol model via Cu-catalyzed dearomatic substitution.12 Most recently, several other related works on remote propargylic substitution have been reported to show the synthetic power of this newly emerging strategy.13 Thus, the development of new catalytic systems for the seldoml studied remote propargylic substitution is highly desired.
We envisioned that with F-containing malonate as the nucleophile, the unprecedented asymmetric remote propargylic monofluoroalkylation might be feasible. However, this proposal is not straightforward. First, a tertiary carbon nucleophile might be less reactive than the widely adopted secondary and primary carbon centers, due to the increased steric hindrance of the former. Meanwhile, α-fluoro carbonyl compounds are known to be less stable than the non-fluorinated ones.14,15 In addition, the elevated acidity of the carbon center in the fluoro malonate nucleophile would also lower the corresponding nucleophilicity and might inhibit the expected substitution process.
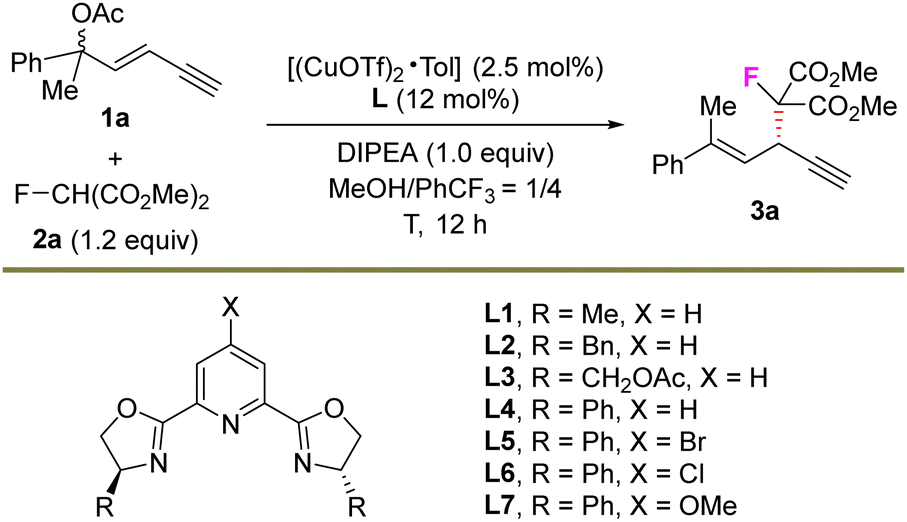
We initiated the study by using 1,3-enyne 1a bearing a tertiary OAc unit as the electrophile, fluorinated malonate 2a as the nucleophile and DIPEA as the base under copper catalysis (Table 1). A series of chiral PyBOx ligands were first evaluated (entries 1–7), and L4 exhibited the highest enantiocontrol, providing SN2′ substitution product 3a in 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 er but with only a 24% yield (entry 4). However, the elevation of the amount of nucleophile 2a greatly increased the yield of 3a to 63% (entry 8). Next, various copper sources were checked but all failed to furnish 3a in a higher yield and stereoselectivity (entries 9–12). When the reaction temperature was lowered to 0 °C with an elongated reaction time, both the yield and enantioselectivity were increased slightly (entry 13). Finally, the optimal reaction conditions were determined as the combination of 1,3-enyne 1a (1.0 equiv.) and fluoro malonate 2a (2.5 equiv.) as the substrates, [(CuOTf)2·Tol]/L4 as the catalyst, and DIPEA as the base in MeOH/PhCF3 as the mixed solvent at −20 °C for 60 h. In this case, 3a was prepared in 82% yield, >20:1 rr, >20:1 E:Z and 98:2 er (entry 14).
:3 er but with only a 24% yield (entry 4). However, the elevation of the amount of nucleophile 2a greatly increased the yield of 3a to 63% (entry 8). Next, various copper sources were checked but all failed to furnish 3a in a higher yield and stereoselectivity (entries 9–12). When the reaction temperature was lowered to 0 °C with an elongated reaction time, both the yield and enantioselectivity were increased slightly (entry 13). Finally, the optimal reaction conditions were determined as the combination of 1,3-enyne 1a (1.0 equiv.) and fluoro malonate 2a (2.5 equiv.) as the substrates, [(CuOTf)2·Tol]/L4 as the catalyst, and DIPEA as the base in MeOH/PhCF3 as the mixed solvent at −20 °C for 60 h. In this case, 3a was prepared in 82% yield, >20:1 rr, >20:1 E:Z and 98:2 er (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | L | T/°C | Yielda (%) | erb |
| a The yield was determined by 1H NMR with CH2Br2 as an internal standard. b Determined by HPLC analysis. c 2a (2.5 equiv.) was used. d CuI (5 mol%) was used instead. e CuCN (5 mol%) was used instead. f [Cu(MeCN)4]BF4 (5 mol%) was used instead. g Cu(OTf)2 (5 mol%) was used instead. h The reaction time was 24 h. i Isolated yield and the reaction time was 60 h. | ||||
| 1 | L1 | RT | 30 | 95:5 |
| 2 | L2 | RT | 24 | 68:32 |
| 3 | L3 | RT | 23 | 82:18 |
| 4 | L4 | RT | 24 | 97:3 |
| 5 | L5 | RT | 28 | 95:5 |
| 6 | L6 | RT | 30 | 94:6 |
| 7 | L7 | RT | 29 | 75:25 |
| 8c | L4 | RT | 63 | 96:4 |
| 9c,d | L4 | RT | 44 | 95:5 |
| 10c,e | L4 | RT | 49 | 96:4 |
| 11c,f | L4 | RT | 58 | 96:4 |
| 12c,g | L4 | RT | 55 | 96:4 |
| 13c,h | L4 | 0 | 70 | 97:3 |
| 14c,i | L4 | −20 | 82 | 98:2 |
With the established protocol in hand, the scope for the asymmetric alkynylallylic monofluoroalkylation reaction was evaluated and the results are summarized in Scheme 2. The enynes bearing various substituted arenes exhibited high compatibility with the transformation. For example, the electrophiles containing F, ether, cyano, CF3, Cl, Br, ester, OCF3 units etc. in the aryl group proceeded smoothly with the stereoselective substitution, affording fluorinated 1,4-enynes (3a–3j, 3l–3n) in 56–89% yields, 96:4–>99:1 er, and generally >20:1 rr and >20:1 E:Z. It should be noted that the aryl iodide motif, which is known to be sensitive to transition metals, was also well tolerated in this process, and the corresponding 3k was formed in 70% yield and 98:2 er, highlighting the broad application scope of the protocol. In addition, other modifications of the substituent in the olefin groups or the nucleophiles did not show obvious erosion of the efficiency and stereocontrol (3o, 3p, and 3r). Alkyl-substituted 1,4-enyne product 3q was also obtained in a similarly good yield and stereoselectivity.
 | ||
| Scheme 2 Scope for the asymmetric alkynylallylic monofluoroalkylation. Isolated yields. a120 h. | ||
Next, we continued to explore the feasibility of the undeveloped asymmetric propargylic monofluoroalkylation reaction (Scheme 3). With the use of L11 instead of L4 as the chiral ligand, the transformation proceeded smoothly, providing 5a in an 84% yield and 94:6 er (see ESI† for the detailed optimization process). A series of substituted aryl-derived alkynes were further evaluated and all showed high compatibility with the reaction. For example, substituents including Cl, Br, I, CF3, cyclopropyl, F, an ether unit etc. in the electrophile reacted with 2a well and generated the corresponding products 5b–5j in 50–80% yields and with 92:8–94:6 er. In addition, the trial to prepare a quaternary stereocenter failed, presumably due to the high steric hindrance for the construction of vicinal quaternary carbon centers.
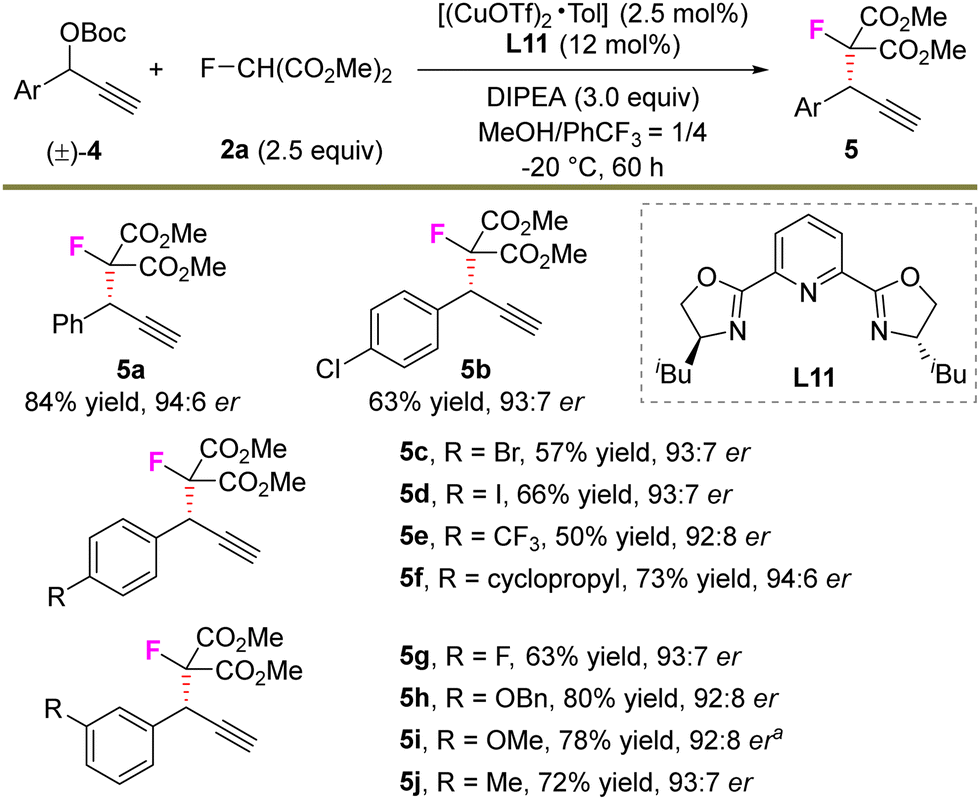 | ||
| Scheme 3 Asymmetric propargylic monofluoroalkylations. Isolated yields. The er values were determined by HPLC analysis. aWhen 1.0 gram of electrophile was used, 1.0 gram of 5i was obtained (88% yield, 92:8 er). | ||
To highlight the robustness and practical use of the present protocol, a gram-scale test was carried out (Scheme 4(a)). When 5.6 mmol of racemic 1g was used, 3g was prepared in 1.4 g in a 76% yield, 97:3 er, >20:1 rr and >20:1 E:Z, comparable to that in 0.1 mmol scale. A set of downstream transformations of 3g were easily conducted and various chiral skeletons were obtained efficiently with high enantioselectivity (6–9). For example, enantioenriched fluoroalkyl-tethered isoxazole 8 was conveniently prepared from 3gvia [3+2] cyclization in 79% yield and 96:4 er. The absolute configuration of 5b was determined to be R by the conversion of 5b to a known compound 10via controlled hydrogenation.16
 | ||
| Scheme 4 Gram-scale test and transformations. | ||
To probe the possible reaction mechanism, nonlinear relationship experiments were conducted (Scheme 5(a)) and a negative nonlinear effect was observed, indicating that multiple ligands might be involved in the enantio-determining step and the heterochiral metal–ligand complex might be more reactive than the homochiral combination.17 Kinetic studies showed that the reaction was first order on the copper catalyst (Scheme 5(b)), suggesting that a monocopper catalyst might be involved in the rate-limiting step.
 | ||
| Scheme 5 Proposed mechanism. | ||
These facts indicated that the observed nonlinear effect might arise from the existence of both an inactive homo dimer of ligands and active mono-Cu(L4) species in the enantio-determining step. Based on this fact and prior work,9 a potential mechanism is described in Scheme 5(c). The copper catalyst reacted with the terminal alkyne first to provide alkynyl copper complex int-1, which was converted to the critical electrophilic olefin-conjugated Cu-allenylidene intermediate int-3 and other tautomers int-2 and int-4. A subsequent nucleophilic attack then occurred on int-3 by 2a to provide fluorinated 1,4-enyne 3a and regenerate the catalyst.
In conclusion, the first copper-catalyzed asymmetric monofluoroalkylation protocol was developed via alkynylallylic substitutions. The related propargylic monofluoroalkylation process is also established. A series of optically active 1,4-enynes bearing a fluoroalkyl unit were prepared in high yields, good regio- and enantioselectivity and excellent E/Z selectivity. The products were conveniently transformed into various privileged chiral skeletons. The preliminary mechanistic studies uncovered a rare negative nonlinear effect.
We acknowledge the National Natural Science Foundation of China (22371292, 22071262), Natural Science Foundation of Ningbo (2023J036), Shanghai Municipal Committee of Science and Technology (22ZR1475200), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0610000), State Key Laboratory of Organometallic Chemistry, and Shanghai Institute of Organic Chemistry for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews on Cu-catalyzed asymmetric propargylic substitutions, see: (a) Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342–356 Search PubMed; (b) R. J. Detz, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2009, 6263–6276 Search PubMed; (c) D.-Y. Zhang and X.-P. Hu, Tetrahedron Lett., 2015, 56, 283–295 Search PubMed; (d) A. F. Adeleke, A. P. N. Brown, L.-J. Cheng, K. A. M. Mosleh and C. J. Cordier, Synthesis, 2017, 790–801 Search PubMed; (e) S. W. Roh, K. Choi and C. Lee, Chem. Rev., 2019, 119, 4293–4356 Search PubMed; (f) Y. Nishibayashi, Chem. Lett., 2021, 50, 1282–1288 Search PubMed; (g) Y.-P. Zhang, Y. You, J.-Q. Yin, Z.-H. Wang, J.-Q. Zhao, Q. Li and W.-C. Yuan, Eur. J. Org. Chem., 2023, e202300728 Search PubMed; (h) Y. You, Y.-P. Zhang, Z.-H. Wang, J.-Q. Zhao, J.-Q. Yin and W.-C. Yuan, Chem. Commun., 2023, 59, 7483–7505 Search PubMed.
- (a) R. J. Detz, M. M. E. H. Delville, H. Hiemstra and J. H. van Maarseveen, Angew. Chem., Int. Ed., 2008, 47, 3777–3780 Search PubMed; (b) G. Hattori, H. Matsuzawa, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2008, 47, 3781–3783 Search PubMed; (c) G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2010, 132, 10592–10608 Search PubMed; (d) C. Zhang, X.-H. Hu, Y.-H. Wang, Z. Zheng, J. Xu and X.-P. Hu, J. Am. Chem. Soc., 2012, 134, 9585–9588 Search PubMed; (e) F.-L. Zhu, Y. Zou, D.-Y. Zhang, Y.-H. Wang, X.-H. Hu, S. Chen, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 1410–1414 Search PubMed; (f) K. Nakajima, M. Shibata and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 2472–2475 Search PubMed; (g) W. Shao, H. Li, C. Liu, C.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 7684–7687 Search PubMed; (h) Q. Wang, T.-R. Li, L.-Q. Lu, M.-M. Li, K. Zhang and W.-J. Xiao, J. Am. Chem. Soc., 2016, 138, 8360–8363 Search PubMed; (i) K. Tsuchida, Y. Senda, K. Nakajima and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 9728–9732 Search PubMed; (j) J. E. Gómez, W. Guo, S. Gaspa and A. W. Kleij, Angew. Chem., Int. Ed., 2017, 56, 15035–15038 Search PubMed; (k) H. Xu, L. Laraia, L. Schneider, K. Louven, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 11232–11236 Search PubMed; (l) K. Zhang, L.-Q. Lu, S. Yao, J.-R. Chen, D.-Q. Shi and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 12847–12854 Search PubMed; (m) R.-Z. Li, H. Tang, L. Wan, X. Zhang, Z. Fu, J. Liu, S. Yang, D. Jia and D. Nius, Chem, 2017, 3, 834–845 Search PubMed; (n) J. Song, Z.-J. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 5212–5216 Search PubMed; (o) R.-Z. Li, H. Tang, K. R. Yang, L.-Q. Wan, X. Zhang, J. Liu, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2017, 56, 7213–7217 Search PubMed; (p) Z.-J. Zhang, L. Zhang, R.-L. Geng, J. Song, X.-H. Chen and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 12190–12194 Search PubMed; (q) J. E. Gómez, À. Cristòfol and A. W. Kleij, Angew. Chem., Int. Ed., 2019, 58, 3903–3907 Search PubMed; (r) R.-Z. Li, D.-Q. Liu and D. Niu, Nat. Catal., 2020, 3, 672–680 Search PubMed; (s) W. Guo, L. Zuo, M. Cui, B. Yan and S. Ni, J. Am. Chem. Soc., 2021, 143, 7629–7634 Search PubMed; (t) J.-T. Xia, L. Li and X.-P. Hu, ACS Catal., 2021, 11, 11843–11848 Search PubMed; (u) H.-D. Qian, Z.-H. Li, S. Deng, C. Yao, H.-M. Xiang, G. Xu, Z.-Q. Geng, Z. Wang, L. Chen, C. Liu, C. Zhu, X. Qi and H. Xu, J. Am. Chem. Soc., 2022, 144, 15779–15785 Search PubMed; (v) F. Gong, X. Meng, S. Lan, J. Liu, S. Yang and X. Fang, ACS Catal., 2022, 12, 12036–12044 Search PubMed; (w) B.-C. Wang, T. Fan, F.-Y. Xiong, P. Chen, K.-X. Fang, Y. Tan, L.-Q. Lu and W.-J. Xiao, J. Am. Chem. Soc., 2022, 144, 19932–19941 Search PubMed; (x) X. Pu, Q.-D. Dang, L. Yang, X. Zhang and D. Niu, Nat. Commun., 2022, 13, 2457 Search PubMed; (y) A. Garcia-Roca, R. Pérez-Soto, G. Stoica, J. Benet-Buchholz, F. Maseras and A. W. Kleij, J. Am. Chem. Soc., 2023, 145, 6442–6452 Search PubMed.
- (a) I. M. Jacobson, S. C. Gordon, K. V. Kowdley, E. M. Yoshida, M. Rodriguez-Torres, M. S. Sulkowski, M. L. Shiffman, E. Lawitz, G. Everson, M. Bennett, E. Schiff, M. T. Al-Assi, G. M. Subramanian, D. An, M. Lin, J. McNally, D. Brainard, W. T. Symonds, J. G. McHutchison, K. Patel, J. Feld, S. Pianko and D. R. Nelson, N. Engl. J. Med., 2013, 368, 1867–1877 Search PubMed; (b) H. Su, Y. Xie, W.-B. Liu and S.-L. You, Bioorg. Med. Chem. Lett., 2011, 21, 3578–3582 Search PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2007, 37, 320–330 Search PubMed; (d) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 Search PubMed.
- X. Gao, Y.-L. Xiao, X.-L. Wan and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3187–3191 Search PubMed.
- X. Gao, R. Cheng, Y.-L. Xiao, X.-L. Wan and X. Zhang, Chem., 2019, 5, 2987–2999 Search PubMed.
- X. Gao, Y.-L. Xiao, S. Zhang, J. Wu and X. Zhang, CCS Chem., 2021, 3, 1463–1471 Search PubMed.
- Y. Hou, Z. Zhang, X. Sun, Z. Yang, Y.-X. Luan and P. Tang, Angew. Chem., Int. Ed., 2023, 62, e202218919 Search PubMed.
- S. Niu, Y. Luo, C. Xu, J. Liu, S. Yang and X. Fang, ACS Catal., 2022, 12, 6840–6850 Search PubMed.
- (a) J.-S. Ma, H.-Y. Lu, Y.-W. Chen, W.-C. Zhao, Y.-Z. Sun, R.-P. Li, H.-X. Wang, G.-Q. Lin and Z.-T. He, Nat. Synth., 2023, 2, 37–48 Search PubMed; (b) J. H. van Maarseveen, Nat. Synth., 2023, 2, 11–12 Search PubMed.
- H.-Y. Lu and Z.-T. He, Chin. Chem. Lett., 2023, 34, 108105 Search PubMed.
- H.-H. Kong, C. Zhu, S. Deng, G. Xu, R. Zhao, C. Yao, H.-M. Xiang, C. Zhao, X. Qi and H. Xu, J. Am. Chem. Soc., 2022, 144, 21347–21355 Search PubMed.
- Y.-Z. Sun, Z.-Y. Ren, Y.-X. Yang, Y. Liu, G.-Q. Lin and Z.-T. He, Angew. Chem., Int. Ed., 2023, 62, e202314517 Search PubMed.
- (a) M.-D. Li, Z.-H. Wang, H. Zhu, X.-R. Wang, J.-R. Wang and T.-Y. Lin, Angew. Chem., Int. Ed., 2023, 62, e202313911 Search PubMed; (b) S.-Y. Luo, G.-Q. Lin and Z.-T. He, Org. Chem. Front., 2024, 11, 690–695 Search PubMed; (c) H.-D. Qian, X. Li, T. Yin, W.-F. Qian, C. Zhao, C. Zhu and H. Xu, Sci. China: Chem., 2024 DOI:10.1007/s11426-023-1922-5.
- Z. Jiao, J. J. Beiger, Y. Jin, S. Ge, J. R. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15980–15986 Search PubMed.
- Z.-T. He, X. Jiang and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 13066–13073 Search PubMed.
- N. Gao, X.-M. Zhao, C.-S. Cai and J.-W. Cai, Org. Biomol. Chem., 2015, 13, 9551–9558 Search PubMed.
- T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456–494 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data and copies of NMR spectra for all products. See DOI: https://doi.org/10.1039/d4cc00371c |
| This journal is © The Royal Society of Chemistry 2024 |