
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn organometallic swap strategy for bottlebrush polymer–protein conjugate synthesis†
Bin
Liu
 ab,
Jacob
Rodriguez
a,
Landon
J. Kilgallon
a,
Wencong
Wang
a,
Yuyan
Wang
a,
Aiden
Wang
a,
Yutong
Dai
a,
Hung V.-T.
Nguyen
a,
Bradley L.
Pentelute
abc and
Jeremiah A.
Johnson
*abc
ab,
Jacob
Rodriguez
a,
Landon
J. Kilgallon
a,
Wencong
Wang
a,
Yuyan
Wang
a,
Aiden
Wang
a,
Yutong
Dai
a,
Hung V.-T.
Nguyen
a,
Bradley L.
Pentelute
abc and
Jeremiah A.
Johnson
*abc
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02142, USA. E-mail: jaj2109@mit.edu
bKoch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02142, USA
cBroad Institute of MIT and Harvard, Massachusetts Institute of Technology Cambridge, MA 02142, USA
First published on 18th March 2024
Abstract
Polymer–protein bioconjugation offers a powerful strategy to alter the physical properties of proteins, and various synthetic polymer compositions and architectures have been investigated for this purpose. Nevertheless, conjugation of molecular bottlebrush polymers (BPs) to proteins remains an unsolved challenge due to the large size of BPs and a general lack of methods to transform the chain ends of BPs into functional groups suitable for bioconjugation. Here, we present a strategy to address this challenge in the context of BPs prepared by “graft-through” ring-opening metathesis polymerization (ROMP), one of the most powerful methods for BP synthesis. Quenching ROMP of PEGylated norbornene macromonomers with an activated enyne terminator facilitates the transformation of the BP Ru alkylidene chain ends into Pd oxidative addition complexes (OACs) for facile bioconjugation. This strategy is shown to be effective for the synthesis of two BP–protein conjugates (albumin and ERG), setting the stage for a new class of BP–protein conjugates for future therapeutic and imaging applications.
Protein-polymer conjugates have garnered significant interest due to their applications in materials science and medicine.1 While a wide range of polymers have been utilized for protein conjugation,1 polyethylene glycol (PEG) is the most extensively studied and translationally successful, with over 25 “PEGylated” proteins approved by the United States Food and Drug Administration (FDA) to date.2 PEGylation can improve the biophysical properties of proteins in several ways.3 For example, the increased size of PEGylated proteins can reduce their renal clearance rate and, as a consequence, increase their circulation half-life.2c–g,3 Additionally, PEGylation can protect proteins from rapid degradation, improve their solubility, and augment their bioavailability and therapeutic efficacy.
Most PEGylated proteins rely on linear PEG macromolecules (Fig. 1a), where features such as molecular weight, number of conjugated PEG chains per protein, and PEG conformation are key variables that impact the biological performance of the resulting conjugates.2c,f,g,3 “Branched” PEGs have also been explored for protein conjugation, where features such as number of branches and branch length can be used to further vary the PEG conformation and physical properties of the resulting conjugates (Fig. 1a).4 For example, Chilkoti and others have utilized polyacrylates with oligoethylene glycol sidechains (∼8–9 ethylene glycol repeat units) for protein conjugation (Fig. 1a). These constructs display many of the desirable properties of linear PEGylated proteins while showing reduced immunogenicity in preclinical studies.5
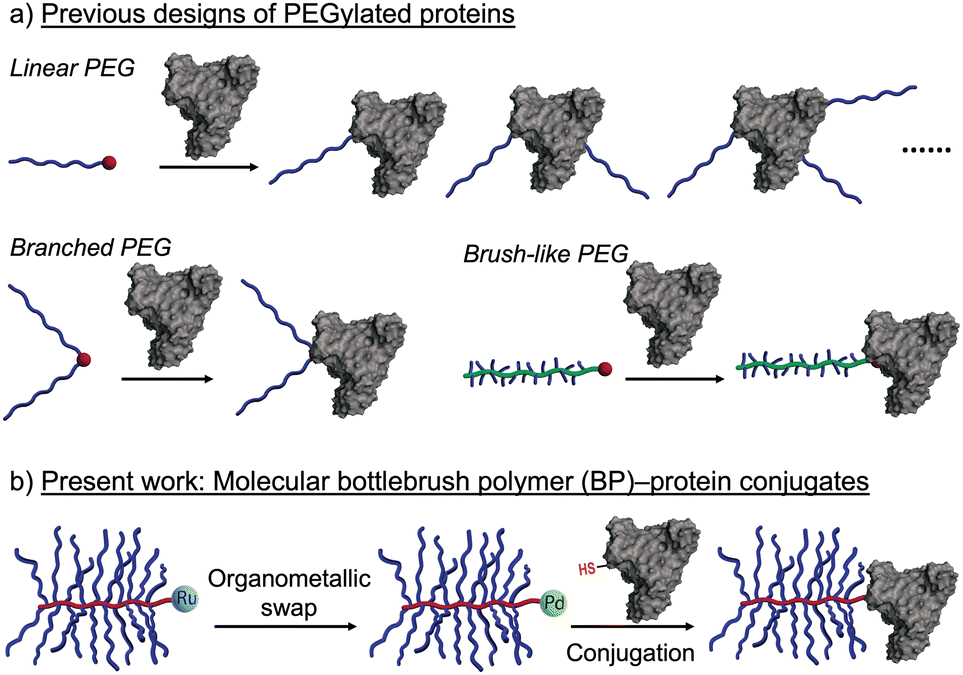 | ||
| Fig. 1 (a) Previous designs of PEGylated proteins with linear, branched, or brush-like architectures. The red dots represent reactive moieties on the polymers used for conjugation. (b) This work introduces molecular bottlebrush polymer (BP)–protein conjugates prepared through a BP chain-end organometallic swap strategy. | ||
These results, along with observations of reduced anti-PEG antibody binding to densely-grafted PEGylated surfaces, suggest that PEGylated molecular bottlebrush polymers (BPs), which feature long PEG side chains bound to a rigid polymer backbone, could be advantageous architectures for protein conjugates. To date, however, such conjugates have not been widely reported, presumably due to synthetic challenges associated with the coupling of large molecules (e.g., proteins and BPs) and incompatibility between the common methods for BP synthesis and bioconjugation. Indeed, PEGylated BPs are perhaps most easily synthesized by “graft-through” ring-opening metathesis polymerization (ROMP), wherein a metal-alkylidene chain end (often Ru-based) reacts with strained olefin macromonomers to propagate chain growth.6 While highly efficient, such metathesis reactions are generally not commensurate with endogenous protein functionality such as amino and thiol amino acid sidechains,7 and efficient methods for conversion of Ru chain ends of BPs to functional groups capable of bioconjugation are not currently available.
Recently, Buchwald, Pentelute, and coworkers pioneered the development of Pd(II) oxidative addition complexes (OACs) as exceptionally efficient reagents for bioconjugation using native cysteine residues of peptides and proteins.8 This method features larger second-order rate constants than most other bio-conjugation reactions, making it particularly suitable for intermolecular coupling of large macromolecules, including protein–protein conjugation. Thus, given that the hydrodynamic sizes of PEGylated BPs prepared by ROMP are similar to those of many proteins (∼5–15 nm), we hypothesized that the challenge of BP–protein conjugation could be solved by the development of a method to quantitatively convert the Ru chains ends of BPs to Pd(II) OACs (Fig. 1b).
Here, we report the realization of this concept. First, we show that it is possible to terminate graft-through ROMP of PEG-based MMs with an enyne derivative featuring a Boc-protected primary amine. Boc-removal and coupling with a Pd(II) OAC containing an N-hydroxysuccinimidyl ester (NHS) provides BP-OACs capable of direct conjugation to cysteine residues of proteins, using bovine serum albumin (BSA) as a model. Conjugation is confirmed by gel electrophoresis and Förster resonance energy transfer (FRET) experiments, while the stability of the aryl thioether linkage is demonstrated by comparison to an analogous maleimide conjugate. Finally, the generality of the method is demonstrated using a second protein—ERG.
Gutekunst and coworkers have shown that enyne derivatives are outstanding terminators for ROMP, yielding linear polymers with nearly complete chain end modification.9 Here, we sought to investigate if such terminators could be used in the context of BPs, which present larger steric limitations, and if they could enable subsequent polymer end-group modification for bioconjugation. Thus, we designed compound 1, which contains an enyne for ROMP termination, a Boc-protected amine for further functionalization, and a PEG linker to provide space between the BP backbone and the Boc-amine. 1 (Fig. 2a) was synthesized in 47% overall yield on the 50 mg scale from a previously reported secondary amine9bvia ring-opening of glutaric anhydride and subsequent amidation using a commercially available heterobifunctional PEG containing primary amine and Boc-protected amine ends (see ESI† for details). PEGylated BPs terminated by 1 (“BP-NHBoc”) were synthesized by ROMP as follows: 3 kDa PEG-based MM (PEG-MM, 20 equiv.) was exposed to Grubbs 3rd-generation bis-pyridyl complex (G3, 1 equiv.) at room temperature (RT) for 30 min in THF solvent to give Ru-terminated BPs “BP-Ru”. The reactions were quenched by the addition of 1 (5 equiv.) and stirring for another 2 h at RT. Size exclusion chromatography (SEC) analysis of the crude reaction revealed a highly efficient polymerization, with nearly quantitative conversion of PEG-MM to a unimodal peak corresponding to putative BP-NHBoc (Mn, SEC = 64 kDa, Đ = 1.04) (Fig. 2b). BP-NHBoc was purified by precipitation in cold diethyl ether to remove excess enyne terminator. The 1H NMR spectrum for BP-NHBoc showed a unique resonance for the Boc end group (Fig. 2c and Fig. S1a, ESI†); the ratio of the integral of the backbone olefinic resonances to the Boc resonance was 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, which is consistent with the theoretical backbone degree of polymerization of 20. Together, these results suggest that 1 is an effective terminator for ROMP of PEGylated BPs.
:9, which is consistent with the theoretical backbone degree of polymerization of 20. Together, these results suggest that 1 is an effective terminator for ROMP of PEGylated BPs.
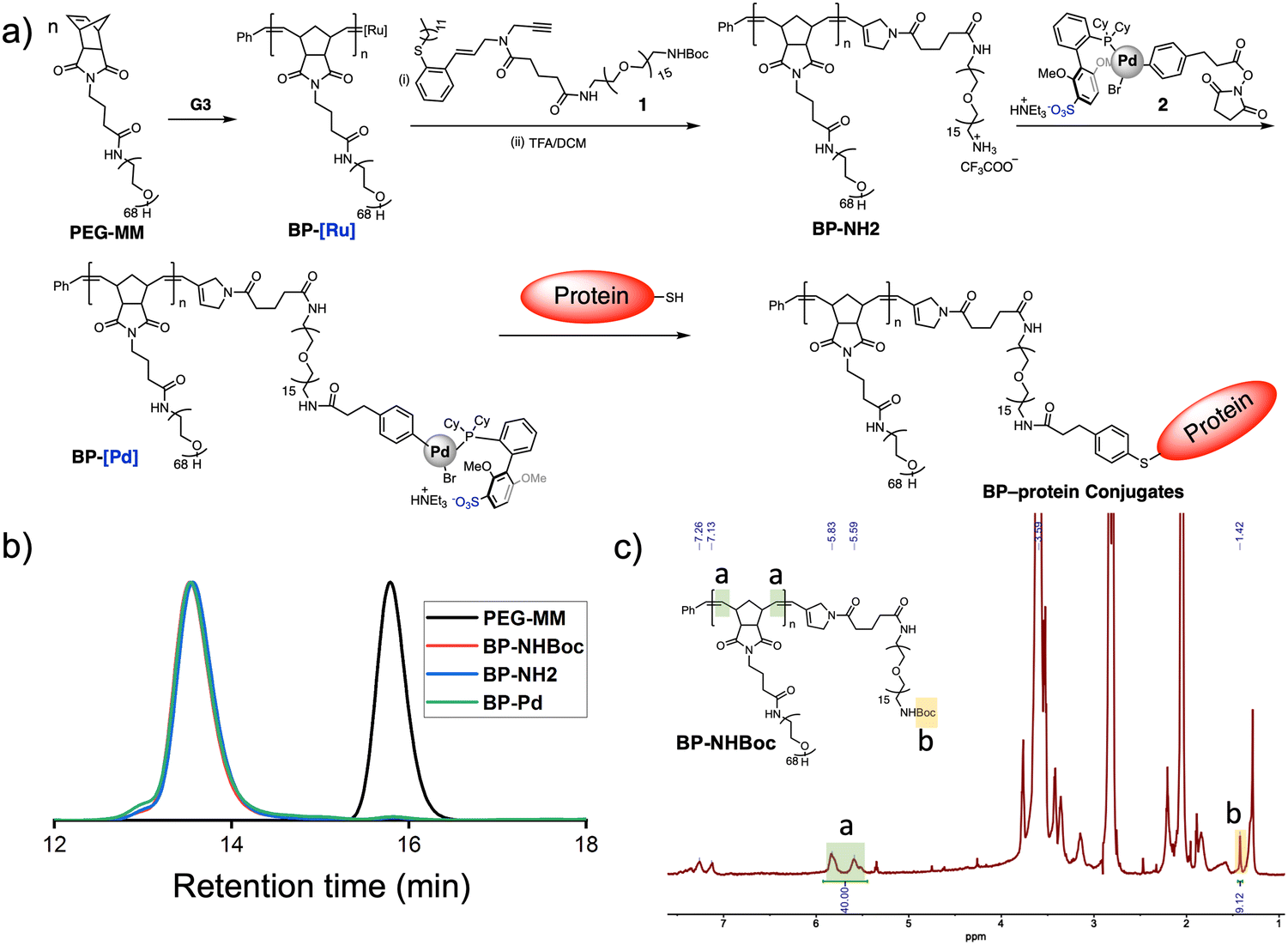 | ||
| Fig. 2 Synthesis of BP-BSA conjugates. (a) Scheme for the synthesis of Pd OAC end-functionalized BPs and BP-BSA. (b) SEC traces of PEG-MM and BPs prior to BSA conjugation. (c) 1H NMR spectrum of BP-NHBoc showing end group fidelity following quenching with enyne 1. | ||
Next, we sought to further modify the end of BP-NHBoc for bioconjugation using Pd(II) OACs. Exposure to trifluoroacetic acid gave complete conversion to the resulting amine-terminated BP BP-NH2 (Fig. S1b, ESI†). Then, mixing with NHS ester 2 (Fig. 2a)8b in phosphate buffer (pH = 8.5) for 12 h gave BP-OAC “BP-Pd”. Inductively coupled plasma-mass spectrometry (ICP-MS) was consistent with nearly quantitative introduction of Pd onto the chain ends (calculated 1000 ppb versus observed 1086 ppb) (Fig. S2, ESI†), with each BP-Pd having one Pd atom. 1H NMR spectroscopy further showed the presence of resonances associated with the OAC (Fig. S3, ESI†), while the size of the BP was conserved as indicated by SEC (Fig. 2b).
With BP-Pd in hand, we explored bioconjugation using BSA (583 amino acids; 66.5 kDa) as a model protein as it contains a single free cysteine available for site-specific conjugation.10 BSA and BP-Pd were combined either in pure water or in phosphate-buffered saline (PBS) (pH = 7.4). Analysis of the crude reaction by SDS-PAGE gel electrophoresis (Fig. 3a and Fig. S4, ESI†) showed a new smear band with a significantly higher molecular weight (∼180 kDa) compared to BSA alone, suggesting the successful formation of conjugate “BP-BSA”. The band at ∼120 kDa for the crude conjugate (Fig. 3a and Fig. S4, ESI†) is assigned to the dimer of BSA. While separation of BP-BSA from free BSA and BP-Pd was difficult using preparatory SEC due to the similar hydrodynamic sizes of each component (Fig. S5, ESI†), anion exchange fast protein liquid chromatography (FPLC) using pH 8.5 buffer (in which BSA is negatively charged; pI = 4.7) enabled facile purification of the conjugates (Fig. 3b) as confirmed by SDS PAGE gel electrophoresis (Fig. 3a). The circular dichroism (CD) spectrum of purified BP-BSA (Fig. 3c) was nearly identical to that of the crude conjugation reaction mixture and BSA alone, suggesting that BSA maintains its folded structure following conjugation and purification.
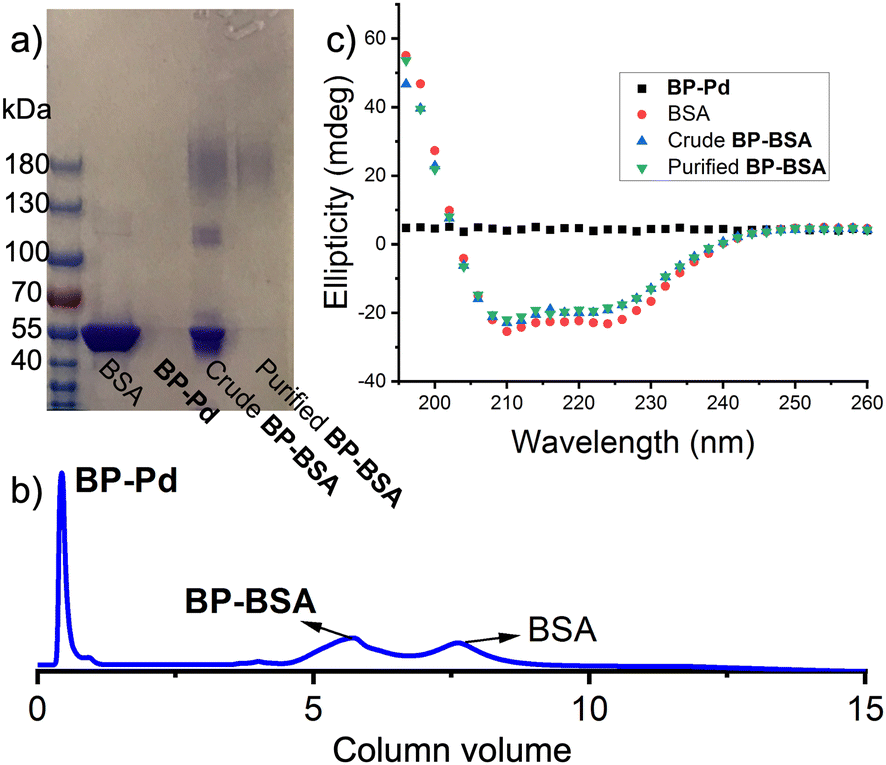 | ||
| Fig. 3 BP-BSA synthesis, purification, and characterization. (a) SDS PAGE Gel showing BSA, BP-Pd, crude BP-BSA, and purified BP-BSA. (b) FPLC trace for crude BP-BSA. (c) CD spectra for BP-Pd, BSA, crude BP-BSA, and purified BP-BSA. | ||
FRET experiments using cyanine 3 (Cy3, λex = 555 nm, λem = 569 nm) and cyanine 5 (Cy5, λex = 651 nm, λem = 670 nm) dyes was designed to provide further evidence for the successful conjugation of BPs to BSA. First, a Cy3-labeled BP “BPCy3-Pd” was synthesized following the same general procedure used for the synthesis of BP-Pd but using a mixture of PEG-MM and 5% of a Cy3-labeled PEG MM (Fig. S6 and S7, ESI†). Meanwhile, Cy5-labeled BSA “BSACy5” was prepared through lysine conjugation using a commercially available Cy-5 NHS ester. Mixing BPCy3-Pd and BSACy5 gave conjugate BPCy3-BSACy5via cysteine arylation. A control conjugate BPCy3-BSA was also prepared using unlabeled BSA. Both conjugates were purified by FPLC as described for BP-BSA (Fig. S8, ESI†). SDS-PAGE gel electrophoresis was used to characterize the proposed conjugates. As expected, BPCy3-BSACy5 showed strong signals in both the Cy3 and Cy5 channels, while BPCy3-BSA and BSACy5 only showed bands in the Cy3 and Cy5 channels, respectively (Fig. 4a). UV-vis spectroscopy supported these findings; the conjugate BPCy3-BSACy5 displayed absorption peaks that overlap with BPCy3-Pd and BSACy5 (Fig. 4b). Fluorescence emission spectra (λex = 550 nm; Cy3 absorption) were collected for BPCy3, BSACy5, a physical mixture of BPCy3 and BSACy5, and FPLC-purified BPCy3-BSACy5. In support of the proposed conjugate structure, FRET-induced Cy5 emission (λem = 670 nm) was only observed for the conjugate BPCy3-BSACy5 (Fig. 4c).
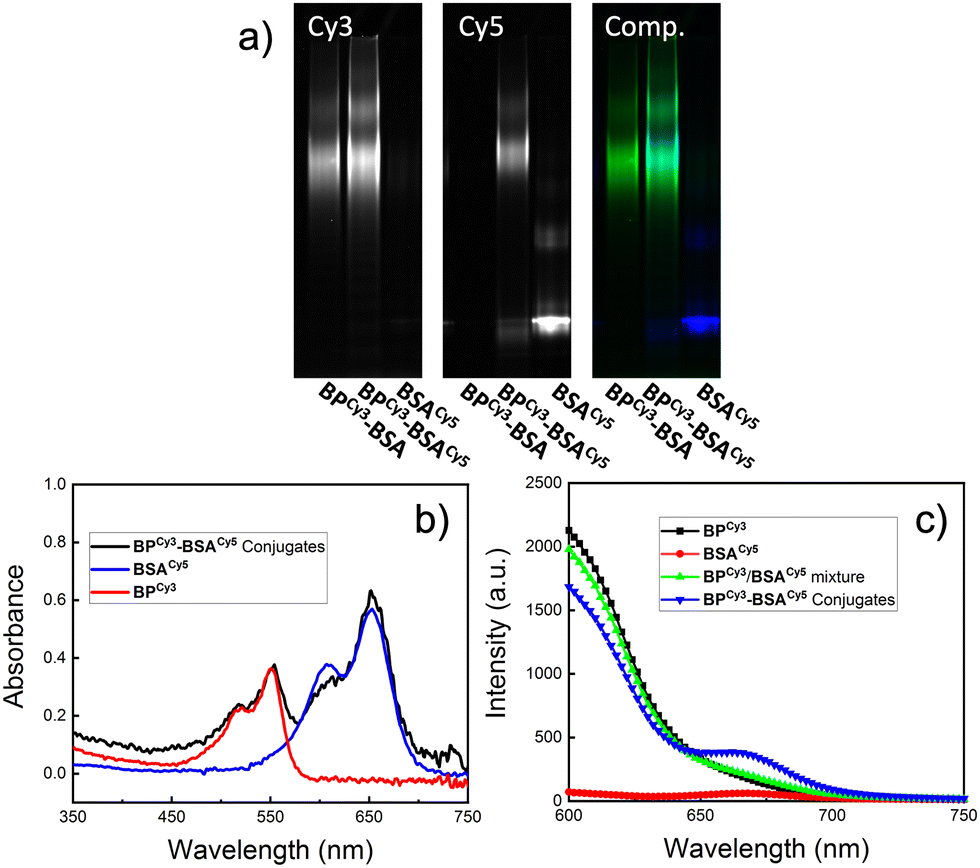 | ||
| Fig. 4 Dye-functionalized BP-BSA conjugates for FRET experiments. (a) Fluorescence gel images under Cy3/Cy5 channels. (b) UV-vis absorbance of BSACy5, BPCy3-Pd, and BPCy3-BSACy5 conjugates. (c) Fluorescence emission spectra for BPCy3-Pd, BSACy5, a physical mixture of BPCy3-Pd and BSACy5, and BPCy3-BSACy5 (λex = 550 nm). | ||
Next, we evaluated the stability of BP-BSA, which features aryl thioether linkages, compared to analogous conjugates prepared using the cysteine-maleimide conjugation reaction (BP-BSA-Mal). Maleimide-terminated BP “BP-Mal” was prepared from BP-NH2 and a commercially available NHS-ester maleimide under similar conditions as were used for the synthesis of BP-Pd from BP-NH2 and 2 (Fig. S9, ESI†), demonstrating generality of our BP end-functionaliazation strategy. Conjugation of BP-Mal to BSA was conducted following commonly reported conditions (in PBS, 24 h). Notably, the conjugation conversion was much lower than for the Pd-mediated arylation reaction, as determined by FPLC (Fig. 3b and Fig. S9c, ESI†). Nevertheless, conjugate BP-BSA-Mal could be isolated following FPLC purification (Fig. S10, ESI†). Exposing BP-BSA and BP-BSA-Mal to glutathione (∼1000 equiv.; 10 mM), which is present in mM concentrations in the cytosol and is known to induce thiolate exchange with thiosuccinimides,11 induced no changes for BP-BSA but led to the emergence of a new SDS-PAGE gel band consistent with free BSA for BP-BSA-Mal. Together, these findings support the successful conjugation of BP and BSA using either Pd(II)-induced cysteine arylation or maleimide conjugation, with the former displaying improved stability toward glutathione.
Finally, to demonstrate the generality of this approach, we formed conjugates between BP-Pd and a chemically synthesized protein, ERG,12 (94 amino acids; ∼11 kDa; Fig. S11a, ESI†) containing a single cysteine residue. Conjugation following the same general procedure used for BSA followed by FPLC purification gave “BP-ERG” in high conversion as indicated by FPLC (Fig. S11b, ESI†) and SDS-PAGE gel electrophoresis (Fig. S11c, ESI†).
In conclusion, we report a strategy to conjugate BPs directly to proteins. Our approach leverages ROMP for BP synthesis and achieves efficient conjugation to cysteine residues of proteins via an “organometallic swap” of Ru to Pd made possible by an enyne ROMP terminator. This approach allows for the merger of two independent methods that are each powerful for the applications in which they were developed (e.g., ROMP for BP synthesis and Pd-mediated cysteine arylation for bioconjugation) but are traditionally orthogonal. The result is a new class of BP–protein conjugates with numerous potential applications that could leverage the utility of BPs for drug delivery and imaging13 with the therapeutic and targeting potential of proteins.
We thank the National Institutes of Health (2R01CA220468-06A1) for support. B. L. thanks the Ludwig Center of the Koch Institute for Integrative Cancer Research at MIT for a postdoctoral fellowship. We thank S. L. Buchwald for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. Chen, D. Y. W. Ng and T. Weil, Prog. Polym. Sci., 2020, 105, 101241 CrossRef CAS; (b) H. G. Börner, Prog. Polym. Sci., 2009, 34, 811–851 CrossRef; (c) A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922 CrossRef CAS; (d) Y. Qi and A. Chilkoti, Curr. Opin. Chem. Biol., 2015, 28, 181 CrossRef CAS PubMed; (e) I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143 CrossRef CAS PubMed; (f) T. A. Wright, R. C. Page and D. Konkolewicz, Polym. Chem., 2019, 10, 434 RSC; (g) M. Takahara, R. Wakabayashi, K. Minamihata, M. Goto and N. Kamiya, Bioconjugate Chem., 2017, 28, 2954 CrossRef CAS PubMed; (h) M. A. Gauthier and H. A. Klok, Chem. Commun., 2008, 2591 RSC; (i) K. Fuhrmann and G. Fuhrmann, Curr. Opin. Colloid Interface Sci., 2017, 31, 67 CrossRef CAS; (j) X. Liu and W. Gao, Angew. Chem., Int. Ed., 2021, 60, 11024 CrossRef CAS PubMed; (k) R. Duncan, Nat. Rev. Cancer, 2006, 6, 688 CrossRef CAS PubMed.
- (a) C. J. White and J. W. Bode, ACS Cent. Sci., 2018, 4, 197 CrossRef CAS PubMed; (b) V. Gupta, S. Bhavanasi, M. Quadir, K. Singh, G. Ghosh, K. Vasamreddy, A. Ghosh, T. J. Siahaan, S. Banerjee and S. K. Banerjee, J. Cell Commun. Signal., 2019, 13, 319 CrossRef CAS PubMed; (c) Y. Hou and H. Lu, Bioconjugate Chem., 2019, 30, 1604 CrossRef CAS PubMed; (d) J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS PubMed; (e) M. Hamidi, A. Azadi and P. Rafiei, Drug Delivery, 2006, 13, 399 CrossRef CAS PubMed; (f) J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170 CrossRef CAS PubMed; (g) M. J. Guicharda, T. Lealb and R. Vanbever, Curr. Opin. Colloid Interface Sci., 2017, 31, 43 CrossRef; (h) https://www.biochempeg.com/article/58.html .
- (a) N. Nischan and C. P. R. Hackenberger, J. Org. Chem., 2014, 79, 10727 CrossRef CAS PubMed; (b) P. B. Lawrence, W. M. Billings, M. B. Miller, B. K. Pandey, A. R. Stephens, M. I. Langlois and J. L. Price, ACS Chem. Biol., 2016, 11, 1805 CrossRef CAS PubMed; (c) J. Morgenstern, P. Baumann and C. Brunner, Int. J. Pharm., 2017, 519, 408 CrossRef CAS PubMed.
- (a) C. Pinholt, J. T. Bukrinsky, S. Hostrup, S. Frokjaer, W. Norde and L. Jorgensen, Eur. J. Pharm. Biopharm., 2011, 77, 139 CrossRef CAS PubMed; (b) J. Ramon, V. Saez, R. Baez, R. Aldana and E. Hardy, Pharm. Res., 2005, 22, 1374 CAS; (c) W. Lee, E. J. Park, S. Kwak, K. C. Lee, D. H. Na and J. S. Bae, Biomacromolecules, 2016, 17, 1160 CrossRef CAS PubMed.
- (a) W. Gao, W. Liu, J. A. Mackay, M. R. Zalutsky, E. J. Toone and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15231 CrossRef CAS PubMed; (b) Y. Qi, A. Simakova, N. J. Ganson, X. Li, K. M. Luginbuhl, I. Ozer, W. Liu, M. S. Hershfield, K. Matyjaszewski and A. Chilkoti, Nat. Biomed. Eng., 2016, 1, 0002 CrossRef PubMed; (c) P. C. Nauka, J. Lee and H. D. Maynard, Polym. Chem., 2016, 7, 2352 RSC.
- (a) O. M. Ogba, N. C. Warner, D. J. O’Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510 RSC; (b) J. Liu, A. O. Burts, Y. Li, A. V. Zhukhovitskiy, M. F. Ottaviani, N. J. Turro and J. A. Johnson, J. Am. Chem. Soc., 2012, 134, 16337 CrossRef CAS PubMed.
- (a) E. Baslé, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213 CrossRef PubMed; (b) O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174 CrossRef CAS PubMed.
- (a) E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687 CrossRef CAS PubMed; (b) H. H. Dhanjee, I. Buslov, I. W. Windsor, R. T. Raines, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2020, 142, 21237 CrossRef CAS PubMed; (c) M. Jbara, J. Rodriguez, H. H. Dhanjee, A. Loas, S. L. Buchwald and B. L. Pentelute, Angew. Chem., Int. Ed., 2021, 60, 12109 CrossRef CAS PubMed.
- (a) T. Zhang, L. Fu and W. R. Gutekunst, Macromolecules, 2018, 51, 6497 CrossRef CAS; (b) L. Fu, T. Zhang, G. Fu and W. R. Gutekunst, J. Am. Chem. Soc., 2018, 140, 12181 CrossRef CAS PubMed.
- J. G. Mehtala, C. Kulczar, M. Lavan, G. Knipp and A. Wei, Bioconjugate Chem., 2015, 26, 941 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946 CrossRef CAS PubMed.
- A. J. Callahan, S. Gandhesiri, T. L. Travaline, R. M. Reja, L. L. Salazar, S. Hanna, Y.-C. Lee, K. Li, O. S. Tokareva, J. M. Swiecicki, A. Loas, G. L. Verdine, J. H. McGee and B. L. Pentelute, Nat. Commun., 2024, 15, 1813 CrossRef CAS PubMed.
- M. Müllner, Chem. Commun., 2022, 58, 5683 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00293h |
| This journal is © The Royal Society of Chemistry 2024 |