
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structure and stereodynamics of atropisomeric N-chloroamides†
Aaron D. G.
Campbell
 a,
Natalie J.
Roper
a,
Paul G.
Waddell
a,
Corinne
Wills
a,
Casey M.
Dixon
a,
Ross M.
Denton
b,
Kristaps
Ermanis
*b and
Roly J.
Armstrong
*a
a,
Natalie J.
Roper
a,
Paul G.
Waddell
a,
Corinne
Wills
a,
Casey M.
Dixon
a,
Ross M.
Denton
b,
Kristaps
Ermanis
*b and
Roly J.
Armstrong
*a
aChemistry – School of Natural and Environmental Sciences, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK. E-mail: roly.armstrong@newcastle.ac.uk
bSchool of Chemistry, University Park, Nottingham, UK. E-mail: kristaps.ermanis@nottingham.ac.uk
First published on 8th March 2024
Abstract
Atropisomeric N-chloroamides were efficiently accessed by electrophilic halogenation of ortho-substituted secondary anilides. The stereodynamics of atropisomerism in these novel scaffolds was interrogated by detailed experimental and computational studies, revealing that racemization is correlated with amide isomerization. The stereoelectronic nature of the amide was shown to significantly influence racemization rates, with potentially important implications for other C–N atropisomeric scaffolds.
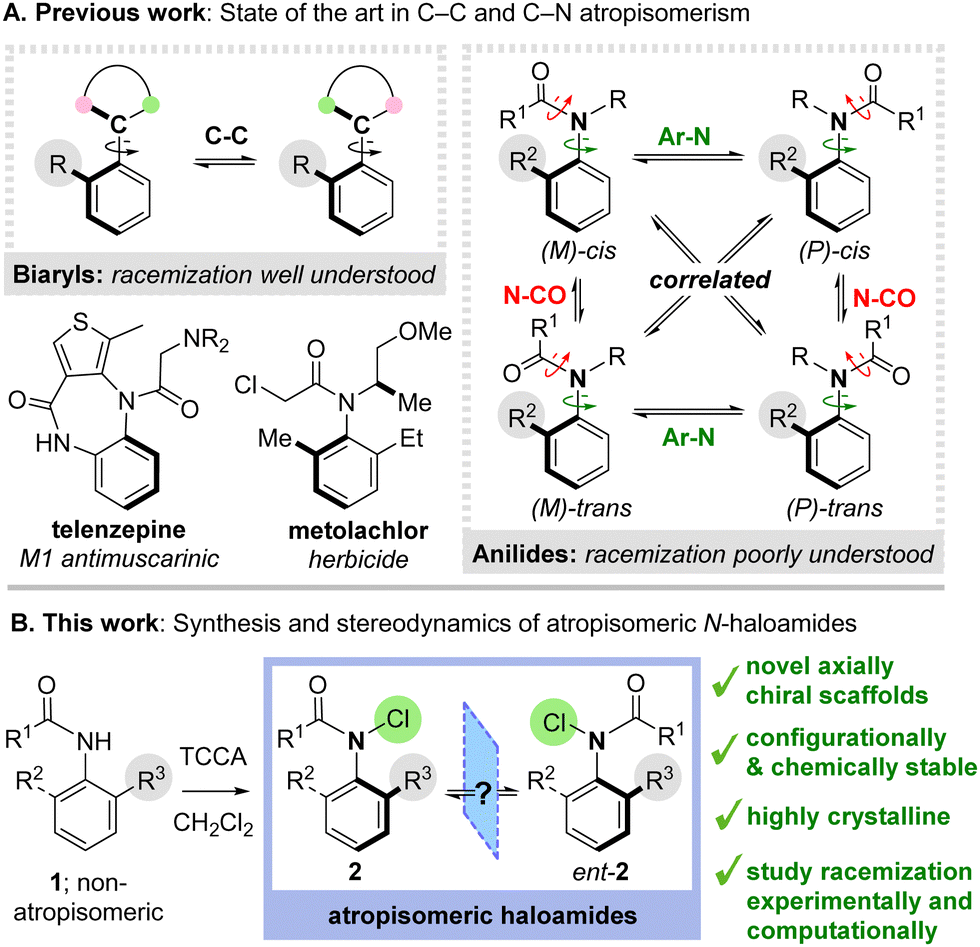
Atropisomers are valuable molecules with powerful applications in catalysis, materials science and medicines.1 The archetypal examples are axially chiral biaryls, whose stereoisomerism derives from restricted rotation about a C–C bond (Scheme 1A).2 The mechanism of racemization in biaryls is well understood and the configurational stability of new materials can be straightforwardly predicted by computational simulation.3 Recently, a growing number of elegant reports have emerged involving atropisomerism about C–N bonds, perhaps most notably in ortho-substituted anilides, which can exhibit robust configurational stability and have emerging potential in medicines (e.g. the drug telenzepine) and agriscience (e.g. the herbicide metolachlor).4 However, in contrast to biaryls, atropisomerism in anilides is complicated by the possibility of rotation about multiple bonds. Although several studies have interrogated the impact of structural modifications on the configurational stability of anilides,5,6 it is not established whether racemization occurs via direct Ar–N bond rotation or in correlation with rotation about the N–CO bond as reported by Clayden and co-workers for C–C atropisomeric benzamides.7 Consequently, it is extremely challenging to predict the racemization rates of new compounds a priori. Understanding the stereodynamic processes underpinning racemization could significantly streamline the identification of new C–N atropisomeric materials.
 | ||
| Scheme 1 Potential and challenges associated with atropisomerism in anilides, and expansion to atropisomeric N-haloamides. | ||
Exemplifying these challenges, we recently questioned whether it might be possible to access unique N-haloamide atropisomers 2via electrophilic chlorination of secondary anilides 1 (Scheme 1B). Given the ubiquitous use of N-haloamide reagents in organic synthesis, accessing the first examples of axially chiral analogues would be of significant interest.8 However, given the unprecedented nature of these scaffolds, it was not clear whether they would display atropisomeric behaviour. Here we report our preliminary results, which reveal that axially chiral N-chloroamides can be efficiently synthesised and display high levels of chemical and configurational stability (trac1/2 up to 12 days). These novel atropisomeric scaffolds have provided an ideal basis to explore the stereodynamics of anilide racemization through experimental and computational studies, providing insight into the degree of correlation with N–CO rotation.
We commenced our study by exploring the electrophilic chlorination of secondary anilides 1 (Scheme 2A). We were pleased to find that treatment with a slight excess of trichloroisocyanuric acid (TCCA) in dichloromethane led to efficient formation of the corresponding N-chloroamides 2, with no evidence of competing aromatic chlorination. This synthetic approach was applied to prepare a library of ortho-substituted chloroamides bearing various sp3 or sp2 acyl substituents (2a–o). Notably, these N-haloamides displayed excellent chemical stability – they could be isolated by routine aqueous workup and column chromatography, characterized using standard spectroscopic methods, and stored at −18 °C for several months without decomposition.
 | ||
| Scheme 2 Synthesis of N-chloroamides and experiments to probe atropisomerism. Yields refer to isolated material after column chromatography. Depictions of absolute stereochemistry are arbitrary. [a] trac1/2 estimated at 20 °C based on ΔG‡ measured at 5 °C by dynamic HPLC analysis. [b] Determined by dynamic HPLC analysis at 20 °C.9,10 | ||
With an efficient synthesis of N-chloroamides established, we investigated their configurational stability. HPLC analysis of 2a on a chiral stationary phase revealed two baseline-separated peaks, consistent with a pair of enantiomers which do not interconvert on the timescale of the HPLC experiment. An enantioenriched sample (obtained by semi-preparative HPLC) was allowed to stand at room temperature and its enantiomeric excess was monitored over time.9 We calculated a racemization half-life (trac1/2) of 12 days corresponding to a Gibbs activation energy of enantiomerization (ΔG‡) of 108.0 kJ mol−1 (see ESI†). Similar analysis was performed on N-chloroamides 2a–o and the resulting values of trac1/2 and ΔG‡ are shown in Scheme 2A.9 Relatively robust configurational stability was observed across analogues bearing various aliphatic acyl substituents (2a–g), but chloroamide 2h bearing a smaller ortho-isopropyl group racemized rapidly.9,10 We also explored anilides 2i–k bearing two ortho substituents. Robust configurational stability was observed for brominated analogue 2k, but chlorinated and fluorinated analogues 2j and 2i underwent comparatively rapid racemization. This is interesting given that C–N atropisomeric sulfonamides derived from 2-chloro-4,6-dimethylaniline have been reported to display higher configurational stability than analogues bearing a single orthotBu-substituent.11 Moreover, examples bearing an aromatic acyl group (2l–n) also racemized extremely rapidly (trac1/2 < 2 h). To investigate this observation further, we plotted the experimentally determined values for ΔG‡ against the Charton steric parameters (ν) associated with the various acyl substituents (Scheme 2B, left).12 This analysis is complicated by the fact that two values of ν are available for the Ph group (out of plane = 0.57; in plane = 1.66), but even for structurally similar aliphatic analogues 2a–g a poor correlation was observed. Shi and co-workers have postulated that electronic effects have an effect on the racemization rate of anilides bearing aromatic acyl substituents, and we speculated that the ability of the amide to deplanarise during racemization may also be playing an important role here.5a We were fortunate to be able to obtain crystals of 2a suitable for single crystal X-ray diffraction allowing us to probe the structure of the amide in the solid state (Scheme 2A). In particular, the planarity of the amide nitrogen atom was studied by evaluating the angle parameter θ, corresponding to the sum of the three valence angles about the nitrogen atom.13 For amide 2a, such analysis revealed a value of θ = 356.6° close to the ideal value of 360° expected from a completely trigonal arrangement. We also obtained X-ray crystal structures of several other N-chloroamides depicted in Scheme 2 (2a–b, d–g, l–n), many of which displayed significantly more distorted nitrogen atoms (e.g.2g, R1 = Cy, θ = 346.1°). Interestingly, when θ was plotted against ΔG‡, for aliphatic examples 2a–g a positive correlation (R2 = 0.90) was observed (Scheme 2B, right). A similar trend was observed for examples 2l–n bearing aromatic amide substituents. This data must be interpreted with some caution, but may suggest that amides possessing a more conformationally distorted ground state are able to racemize more rapidly. However this effect does not explain the significantly reduced configurational stability of examples containing aliphatic vs. aromatic acyl substituents (e.g., anilides 2d and 2l possess similar values of θ, but 2l racemizes two orders of magnitude more rapidly).
A similar effect has been reported by Curran and co-workers in atropisomeric N-methyl anilides, with benzoylated analogues racemizing significantly faster than examples bearing aliphatic acyl substituents.5e In this case, it was also noted that benzoylated analogues displayed a higher preference for the trans-amide isomer. In line with this observation, we noticed that the X-ray structures of aliphatic chloroanilides (2a–b, d–g) possessed a cis-configured amide, but in contrast, the structures of aromatic anilides 2l–n featured a trans-amide geometry.14 To probe if similar behaviour is observed in solution, we analyzed 2a by 1H NMR in CDCl3 at 298 K and observed a single set of broad peaks (Scheme 3A). Upon cooling to 233 K, the spectrum resolved to reveal two well-defined species present in a 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 mixture, which were characterized as the cis- and trans-amide isomers respectively on the basis of NOE-analysis. Strong EXSY correlations were also observed between geometrical isomers, indicating relatively rapid exchange even at 273 K (see ESI† for details). Phenyl-substituted amide 2l also exhibited a single set of broad peaks at room temperature, but upon cooling to 218 K resolved into two geometrical isomers with a slight preference for the trans-isomer (Scheme 3B). From this data, we concluded: (i) geometrical isomerization of the amide is extremely fast compared with racemization; (ii) the ratio of geometrical isomers is governed by thermodynamics and not established during chlorination; (iii) the nature of the acyl substituent exerts a significant influence on the position of equilibrium, with aliphatic groups resulting in a near exclusive preference for the cis-geometry but aromatic groups leading to a higher equilibrium population of the trans-isomer.
:7 mixture, which were characterized as the cis- and trans-amide isomers respectively on the basis of NOE-analysis. Strong EXSY correlations were also observed between geometrical isomers, indicating relatively rapid exchange even at 273 K (see ESI† for details). Phenyl-substituted amide 2l also exhibited a single set of broad peaks at room temperature, but upon cooling to 218 K resolved into two geometrical isomers with a slight preference for the trans-isomer (Scheme 3B). From this data, we concluded: (i) geometrical isomerization of the amide is extremely fast compared with racemization; (ii) the ratio of geometrical isomers is governed by thermodynamics and not established during chlorination; (iii) the nature of the acyl substituent exerts a significant influence on the position of equilibrium, with aliphatic groups resulting in a near exclusive preference for the cis-geometry but aromatic groups leading to a higher equilibrium population of the trans-isomer.
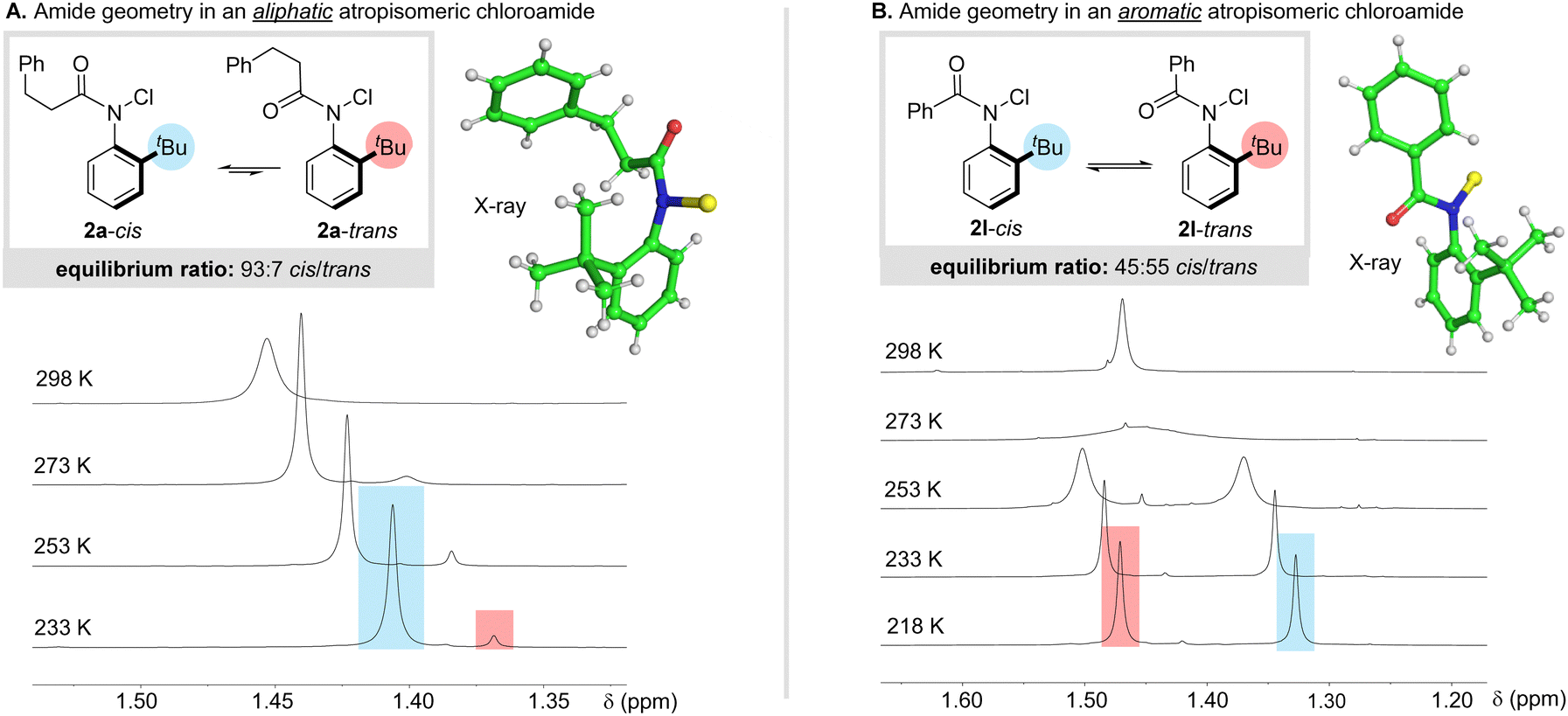 | ||
| Scheme 3 Conformational analysis of aromatic and aliphatic N-chloroamides 2a and 2l by variable-temperature 1H NMR spectroscopy and X-ray crystallography. Stack plots show the tert-butyl region, with spectra offset for clarity (full spectra are included in the ESI†). | ||
To gain insight into how these factors relate to configurational stability, DFT modelling studies were conducted (ωB97M-V15/def2-QZVPP16/SMD(n-hexane)17//PBE0-D318/def2-TZVP/SMD(n-hexane), see ESI† for full details). For a representative chloroamide 2b (Scheme 4A), our analysis suggested that the lowest energy racemization pathway occurs via a Clayden-type correlated mechanism, in which Ar–N bond rotation is geared with N–CO isomerization (calculated ΔG‡ = 117.2 kJ mol−1). We were unable to find any energetically feasible processes involving direct Ar–N bond rotation (without amide isomerization) from either cis- or trans-amide rotamers. (P)-trans-2b formed in this correlated isomerization was calculated to be destabilised with respect to the starting material by 2.1 kJ mol−1, but can isomerize to the thermodynamically favoured cis-amide via a facile N–CO bond rotation (ΔG‡ = 58.7 kJ mol−1). This is broadly consistent with our experimental observations for 2b, namely a cis/trans ratio of ∼9:1 observed at 263 K by 1H NMR, and rapid exchange of amide rotamers at room temperature. Equivalent analyses of chloroamides 2d, 2l and 2n suggested a similar racemization mechanism via correlated Ar–N and N–CO rotation (Scheme 4B). The calculated values of ΔG‡ were somewhat higher than those determined experimentally, but reproduced the experimentally observed trend with good fidelity (2b > 2d ≫ 2l > 2n). For all four examples, the computed racemization pathway involves a significant reduction in N–CO bond order from 1.13 to 0.95 on average, accompanied by a smaller increase in Ar–N bond order in the transition state (TS) from 0.85 to 1.01 on average. In all cases there was also a decrease in the planarity of the N atom in the TS compared to the ground state. This is consistent with deconjugation of the nitrogen lone pair from the amide in order to partially re-conjugate with the ortho-substituted aromatic ring and is also in line with our experimental result that increased distortion about the nitrogen atom in the ground state (i.e. weakening of the amide bond) correlates with more facile racemization (also replicated computationally, see ESI†). In line with the experimental data, for 2l and 2n our calculations also show that the cis-amide is destabilised relative to the trans-isomer by 3.4 and 4.8 kJ mol−1, respectively. This is likely a consequence of an unfavourable interaction in the cis-amide between the benzoyl group and the N–Ar group. Interestingly, in all four compounds the cis isomer features a stronger amide bond than in the trans (bond order 1.17 vs. 1.11 on average). Overall, we believe that the experimentally and computationally observed ground state destabilization in benzoylated analogues may explain their significantly more facile racemization.
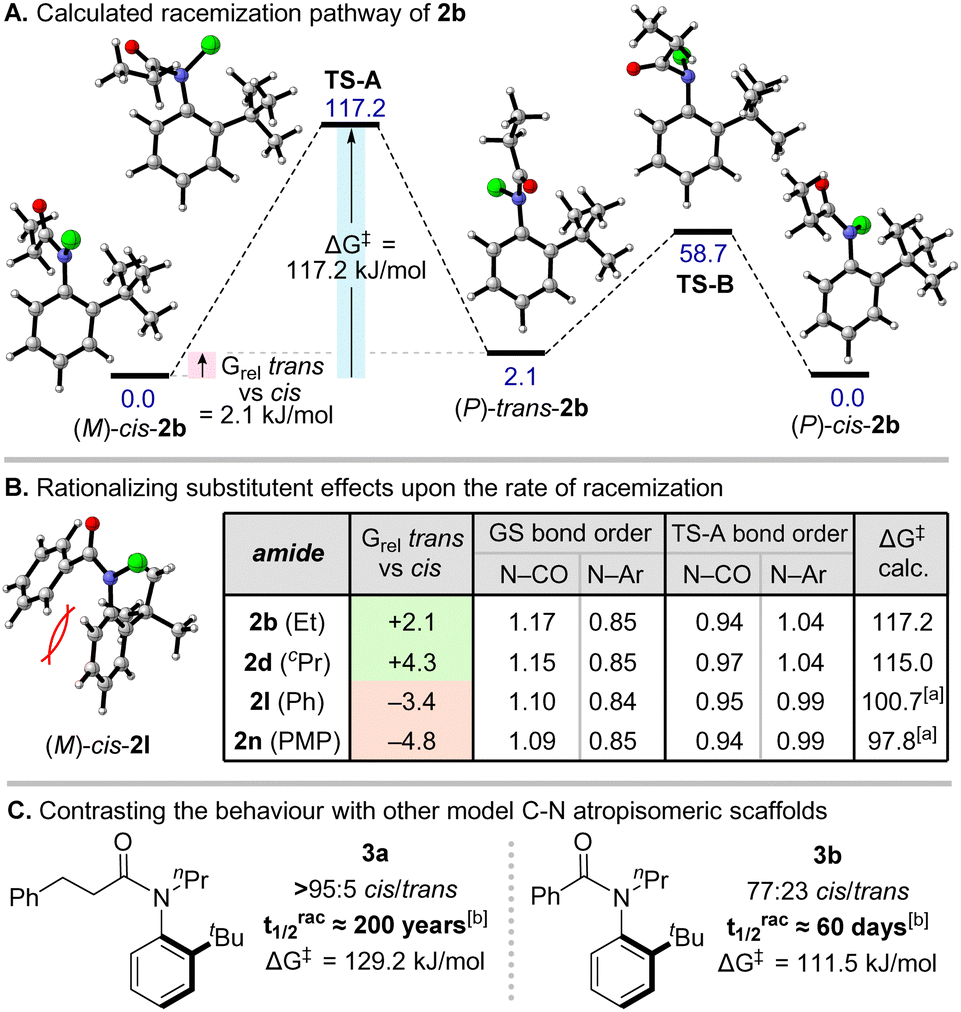 | ||
| Scheme 4 Computational analysis of chloroanilide racemization and comparison with model scaffolds bearing non-halogen N-substituents. Calculations were carried out at the PBE0-D3/def2-tzvp/SMD(n-hexane)//wB97M-V/def2-QZVPP/SMD(n-hexane) level. Free energies are reported in kJ mol−1. [a] ΔG‡ calculated from lowest energy trans-amide. [b] trac1/2 for 3a and 3b estimated at 20 °C based on ΔG‡ measured at elevated temperature in 2,2,4-trimethylpentane (see ESI†). | ||
Finally, to establish the generality of our conclusions, we prepared compounds 3a–b bearing an N-propyl group (Scheme 4C). In line with our study, hydrocinnamoyl analogue 3a was observed as a single cis-amide (>95:5 cis/trans) with a high racemization barrier (trac1/2 ≈ 200 years). In contrast, for benzoyl analogue 3b, an appreciable amount of trans-amide was observed (77:23 cis/trans) accompanied by a dramatic reduction in configurational stability (trac1/2 ≈ 60 days). Taken together with the similar results reported by Curran and co-workers, these results imply that the stereodynamic processes described here may apply more generally to other C–N atropisomers.
In summary, we have developed the first synthesis of atropisomeric N-haloamides. Experimental and computational studies established that stereoelectronics of the amide play a key role in dictating the racemization rate. Computational studies suggest racemization occurs via correlated rotation about the Ar–N and N–CO bonds, which may have important implications for the wider field of C–N atropisomerism. Research in our laboratory is currently ongoing to develop asymmetric syntheses of N-chloroamides and harness them in asymmetric halogenation processes.
We gratefully acknowledge financial support from EPSRC (EP/S022791/1), Newcastle University, the Royal Society (RGS\R1\221162) and the Bill and Milica Beck PhD Endowment Fund. We also thank Vertex Pharmaceuticals for generous provision of equipment. Calculations were performed using the Sulis Tier 2 HPC platform hosted by the Scientific Computing Research Technology Platform at the University of Warwick. Sulis is funded by EPSRC Grant EP/T022108/1 and the HPC Midlands+ consortium.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed.
- J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC.
- S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef CAS PubMed.
- (a) J. S. Sweet and P. C. Knipe, Synthesis, 2022, 2119–2132 CAS; (b) Y.-J. Wu, G. Liao and B.-F. Shi, Green Synth. Catal., 2022, 3, 117–136 CrossRef; (c) P. Rodríguez-Salamanca, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem. – Eur. J., 2022, 28, e202104442 CrossRef PubMed; (d) G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chemistry, 2022, 8, 1855–1893 CrossRef CAS; (e) A. D. G. Campbell and R. J. Armstrong, Synthesis, 2023, 2427–2438 CAS.
- (a) Q.-J. Yao, P.-P. Xie, Y.-J. Wu, Y.-L. Feng, M.-Y. Teng, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2020, 142, 18266–18276 CrossRef CAS PubMed; (b) N. Suzumura, M. Kageyama, D. Kamimura, T. Inagaki, Y. Dobashi, H. Hasegawa, H. Fukaya and O. Kitagawa, Tetrahedron Lett., 2012, 53, 4332–4336 CrossRef CAS; (c) K. Kondo, H. Fujita, T. Suzuki and Y. Murakami, Tetrahedron Lett., 1999, 40, 5577–5580 CrossRef CAS; (d) K. Kondo, T. Iida, H. Fujita, T. Suzuki, K. Yamaguchi and Y. Murakami, Tetrahedron, 2000, 56, 8883–8891 CrossRef CAS; (e) D. P. Curran, G. R. Hale, S. J. Geib, A. Balog, Q. B. Cass, A. L. G. Degani, M. Z. Hernandes and L. C. G. Freitas, Tetrahedron: Asymmetry, 1997, 8, 3955–3975 CrossRef CAS.
- For related studies on amines, see: (a) R. Costil, A. J. Sterling, F. Duarte and J. Clayden, Angew. Chem., Int. Ed., 2020, 59, 18670–18678 CrossRef CAS PubMed; (b) T. Kawabata, C. Jiang, K. Hayashi, K. Tsubaki, T. Yoshimura, S. Majumdar, T. Sasamori and N. Tokitoh, J. Am. Chem. Soc., 2009, 131, 54–55 CrossRef CAS PubMed; (c) Y. Iwasaki, R. Morisawa, S. Yokojima, H. Hasegawa, C. Roussel, N. Vanthuyne, E. Caytan and O. Kitagawa, Chem. – Eur. J., 2018, 24, 4453–4458 CrossRef CAS PubMed; G. Furukawa, T. Shirai, Y. Homma, E. Caytan, N. Vanthuyne, D. Farran, C. Roussel and O. Kitagawa, J. Org. Chem., 2020, 85, 5109–5113 Search PubMed; (d) D. Homma, S. Taketani, T. Shirai, E. Caytan, C. Roussel, J. Elguero, I. Alkorta and O. Kitagawa, J. Org. Chem., 2022, 87, 8118–8125 CrossRef CAS PubMed.
- (a) J. Clayden and J. H. Pink, Angew. Chem., Int. Ed., 1998, 37, 1937–1939 CrossRef CAS; (b) R. A. Bragg, J. Clayden, G. A. Morris and J. H. Pink, Chem. – Eur. J., 2002, 8, 1279–1289 CrossRef CAS PubMed; (c) D. R. Hirsch, A. J. Metrano, E. A. Stone, G. Storch, S. J. Miller and R. P. Murelli, Org. Lett., 2019, 21, 2412–2415 CrossRef CAS PubMed.
- W. M. Gołebiewski and M. Gucma, Synthesis, 2007, 3599–3619 CrossRef.
- J.-P. Heeb, J. Clayden, M. D. Smith and R. J. Armstrong, Nat. Protoc., 2023, 18, 2745–2771 CrossRef CAS PubMed.
- O. Trapp, Chirality, 2006, 18, 489–497 CrossRef CAS PubMed.
- Z. Gao, C.-X. Yan, J. Qian, H. Yang, P. Zhou, J. Zhang and G. Jiang, ACS Catal., 2021, 11, 6931–6938 CrossRef CAS.
- (a) M. Charton, J. Am. Chem. Soc., 1975, 97, 1552–1556 CrossRef CAS; (b) M. Charton, J. Org. Chem., 1976, 41, 2217–2220 CrossRef CAS.
- Y. Otani, O. Nagae, Y. Naruse, S. Inagaki, M. Ohno, K. Yamaguchi, G. Yamamoto, M. Uchiyama and T. Ohwada, J. Am. Chem. Soc., 2003, 125, 15191–15199 CrossRef CAS PubMed.
- G. J. Pros and A. J. Bloomfield, J. Phys. Chem. A, 2019, 123, 7609–7618 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2016, 144, 214110 CrossRef PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data for new compounds. CCDC 2307639–2307647. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc00268g |
| This journal is © The Royal Society of Chemistry 2024 |