
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArbuzov meets 1,2-oxaphosphetanes: transient 1,2-oxaphosphetan-2-iums as an entry point to beta-halo phosphane oxides and P-containing oligomers†‡
Florian
Gleim
a,
Gregor
Schnakenburg
 a,
Arturo Espinosa
Ferao
*b and
Rainer
Streubel
*a
a,
Arturo Espinosa
Ferao
*b and
Rainer
Streubel
*a
aInstitut für Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhardt-Domagk-Straße 1, 53121 Bonn, Germany. E-mail: r.streubel@uni-bonn.de
bFacultad de Química, Campus de Espinardo, Universidad de Murcia, 30100 Murcia, Spain. E-mail: artuesp@um.es
First published on 7th February 2024
Abstract
Herein, we describe the synthesis of a 1,2σ3λ3-oxaphosphetane from ethylene oxide and its reactions with alkyl halides to form β-halo phosphane oxides in an Arbuzov-type reaction. When methyl triflate was used as a hard electrophile, cationic oligomerisation of 1,2-oxaphosphetanes was observed. DFT calculations indicate 1,2-oxaphosphetan-2-iums as intermediates and reveal differences between the Arbuzov and the potential Perkow reaction pathway.
Strained organic and inorganic ring systems1 are highly interesting due to their special bonding situation and high reactivity. For example, oxetanes (I) (Fig. 1) are important building blocks for synthesising more complicated molecules2 and polymers.3 The phosphorus-containing four-membered rings such as phosphetanes (II) drew attention because of their use as steering ligands in transition metal catalysis,4 and, more recently, as organocatalysts.5–10 Oxaphosphetanes (III, IV) represent an unusual combination of oxetanes and phosphetanes but have been scarcely studied, so far. It can be anticipated that 1,3-oxaphosphetanes (III)11 and 1,2-oxaphosphetanes (IV)12 will behave quite differently whereby III should possess features more similar to I and II. Interestingly, higher coordinate derivatives of III and IV have been investigated more often. For example, 1,3σ4λ5-oxaphosphetanes are available through intramolecular Mitsunobu reactions,11 and bi- and tricyclic 1,3σ3λ3-oxaphosphetanes have recently been proposed in the decomposition of HPCO.13 The 1,2σ5λ5-oxaphosphetanes (IVb) were discussed as intermediates in the lithium-free Wittig reaction,12,14,15 and also in the deoxygenation of epoxides.15–17 Recent DFT calculations showed that they also occur in the phosphite-initiated reductive dimerization of ketones.18 Until now, very few crystal structures of 1,2σ5λ5-oxaphosphetanes (IVb) have been reported.19–22 In contrast, the lower coordinate 1,2σ3λ3-oxaphosphetanes (IVa) were only proposed for a long time, with no known stable derivatives.23 In 2016, we reported the synthesis of pentacarbonylmetal(0) complexes (M = Cr, Mo, and W) (V) either through ring expansion of epoxides using highly reactive Li/Cl phosphinidenoid complexes or ring formation through intramolecular nucleophilic attack in a α,ω-bifunctional phosphane complex.24–26 Recently, we also gained access to the free ligand27 (IVa) using thermal decomplexation28 from complexes [M(CO)5L] based on the chelating properties of bis(diphenylphosphino)ethane (DPPE). The first SC-XRD structure of a non-ligated 1,2σ3λ3-oxaphosphetane (IVa) was reported together with examples of P-oxidation reactions using ortho-chloranil and P-complexation by gold(I) chloride.27 Subsequently, we reported on an elusive 1,2σ4λ5-oxaphos-phetane P-oxide (VI) and also on a stable P-sulfide and a P-selenide, formerly unknown.29
 | ||
| Fig. 1 Oxetane (I), phosphetanes (σ3λ3IIa, σ5λ5IIb), 1,3-oxaphosphetanes (σ3λ3IIIa, σ5λ5IIIb), 1,2-oxaphosphetanes (σ3λ3IVa, σ5λ5IVb), 1,2σ3λ3-oxaphosphetane metal complexes (V), 1,2σ4λ5-oxaphosphetanes (VI) and 1,2σ4λ4-oxaphos-phetan-2-ium (VII). | ||
We became attracted to test the usability of 1,2σ3λ3-oxaphosphetanes IVa as a building block for functional organophosphorus compounds via the Arbuzov reaction. The latter is an important and well-studied reaction for acyclic phosphanes,30,31 bearing a P-alkoxy substituent, with alkyl halides, thus rendering functional phosphane oxides as final products (Scheme 1).30,32 Cyclic versions of this reaction have been studied so far only for 1,2-oxaphospholanes, but not for smaller P-heterocyclic rings such as in IVa.33,34
 | ||
| Scheme 1 Examples of Arbuzov reactions for: (a) general, acyclic compounds VIII; (b) 1,2σ3λ3-oxaphosphetanes IVa, presented in this work. | ||
Herein, we report on the synthesis and reactions of 1,2-oxaphosphetanes with various alkyl halides thus rendering a variety of β-halo phosphane oxides XII involving transient 1,2-oxaphosphetan-2-ium species VII as indicated by DFT calculations. Furthermore, preliminary results of a cationic oligo-merization of a 1,2-oxaphosphetane is presented.
To avoid complications from the use of diastereomeric 1,2σ3λ3-oxaphosphetanes, we first synthesized C4-unsubstituted and symmetrically C4-disubstituted 1,2-oxaphosphetane complexes. Li/Cl phosphinidenoid molybdenum complex 1 reacted with oxirane or 1,1′-dimethyloxirane to yield selectively the corresponding 1,2-oxaphosphetane molybdenum(0) complexes 4 (65% yield) and 5 (77% yield) (Scheme 2). To our surprise, the subsequent treatment of toluene solutions of 4 and 5 with DPPE at 80 °C led to partial decomposition in the case of 5, whereas selective formation of unligated 1,2-oxaphosphetane 6 (70% yield) was observed for 4.
 | ||
| Scheme 2 Synthesis of 1,2-oxaphosphetanes molybdenum complexes 4 and 5 and free 1,2-oxaphosphetanes 6 and 7. | ||
The outcome for 5 could be somewhat improved by reducing the reaction time from 4 h to 2 h thus enabling to detect a resonance signal at 161.6 ppm in the 31P-NMR spectrum of the reaction mixture which was assigned to 7 (content estimated 18%, by 31P{1H} NMR integration). Our established protocol enabled us to isolate 1,2-oxaphosphetane 6, but 7 could only be observed as a mixture in solution (Scheme 2).27,29 Complexes 4 and 5 as well as the “free” 1,2-oxaphosphetane 6 were structurally confirmed via single crystal X-ray diffraction (SC-XRD) studies. Interestingly, the structural features of 4 and 6 (Fig. 2) are very similar, i.e., an almost planar ring system and similar bond lengths (see the ESI‡). But the 31P{1H} NMR chemical shifts vary distinctively for both complexes (219.5 ppm (4) and 183.9 ppm (5)) and the unligated 1,2-oxaphosphetanes (212.1 ppm (6) and 161.6 ppm (7)).
 | ||
| Fig. 2 Molecular structures of 4 (left) and 6 (right) in the solid state. Hydrogen atoms are omitted, and the thermal ellipsoids are set at the 50% probability level. For solid state structures of 5 see the ESI.‡ Selected bond lengths [Å] and angles [°]: 4: Mo–P 2.487, P–O 1.661(3), P–C1 1.845(5), P–C3 1.895(5), and C1–P–O 80.81(19). 6: P–O 1.670(3), P–C1 1.849(4), P–C3 1.923(5), and C1–P–O 80.47(18). | ||
To explore the reactivity of 6, it was treated with various activated alkyl halides 8a–d to achieve alkylation at phosphorus to give 1,2-oxaphosphetan-2-ium salts 9a–d as intermediates. As acyclic β-halo phosphane oxides 10a–d were obtained in an Arbuzov-type reaction (Scheme 3), we concluded that a rapid cleavage of the O–C4 bond upon nucleophilic attack of the counter anion had followed the P-alkylation step instead of forming P-functional 1,2σ4λ5-oxaphosphetanes. The reactions of 6 with 8a–d were selective and products 10a–d were isolated in moderate yields (Table 1). For 10a–c SC-XRD measurements (Fig. 3) confirmed all proposed structures.
 | ||
| Scheme 3 Arbuzov reactions of 6 yielding β-halo phosphane oxides 10a–d. | ||
| 10a | 10b | 10c | 10d | |
|---|---|---|---|---|
| δ [ppm] | 48.4 | 49.7 | 48.5 | 53.6 |
| Yield [%] | 42 | 53 | 6 | 57 |
 | ||
| Fig. 3 Molecular structures of 10a (left) and 10c (right) in the solid state. Hydrogen atoms are omitted, and the thermal ellipsoids are set at the 50% probability level. For solid state structures of 10b, see the ESI.‡ Selected bond lengths [Å] and angles [°]: 10a: P–O 1.4841(10), P–C1 1.8254(14), P–C3 1.8290(14), P–C6 1.8809(14), and C1–P–C3 104.05(7). 10c: P–O 1.488(3), P–C1 1.823(4), P–C3 1.838(4), P–C6 1.886(3), and C1–P–C3 106.16(17). | ||
The effect of the counter anion was then examined. While a clean reaction occurred with methyl iodide (8d), leading to 10d, methyl triflate (8e) did not enable the reaction of transiently formed 9e with the counter anion at low temperature, thus behaving as a weakly coordinating anion (WCA).35,36 Therefore, no “monomeric” molecular compound was formed, and the cationic intermediate 9e reacted with further units of 6 in propagation steps to give a mixture of oligomeric compounds “11n” (Scheme 4). Analysis of the solid residue of the reaction mixture by MALDI-MS revealed oligomers with up to ten units of 6 incorporated (m/z 318.1). Repeating the experiment with different equivalents of MeOTf (Fig. 4) showed that, in general, a lesser amount of MeOTf (0.25 eq., 0.05 eq.) led to an increase in heavier oligomers 11,6–10 but no oligomers with a chain length longer than n = 10 were detected. Dimer 112 formation was favoured for 0.5 eq. of reagent, whereas a pentamer 115 was preferred for all other sub-stoichiometric ratios. The largest relative ratio of heavier oligomers 116-10 was observed for 0.05 eq. of MeOTf.
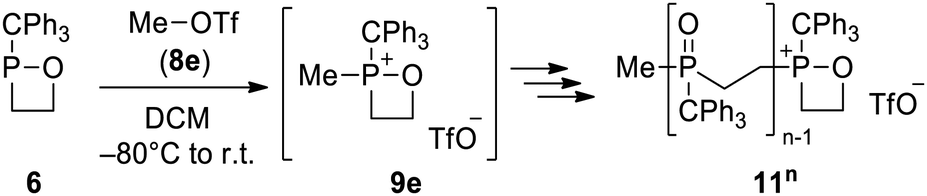 | ||
| Scheme 4 Arbuzov reaction of 6 with methyl triflate leading to a mixture of oligomers 11n (n = 2–10). | ||
 | ||
| Fig. 4 Relative distribution of chain length of oligomers of 11n, depending on equivalents of MeOTf with constant concentration of 6, as detected by MALDI mass spectrometry. | ||
VT-NMR monitoring showed a complex reaction progress. In the 31P{1H}-NMR spectra, a signal at 130.4 ppm was detected between −80 and −20 °C. This signal is tentatively assigned to the 2-methyl-1,2-oxaphosphetan-2-ium salt 9e; this was further supported by the calculated 31P chemical shift of 132.5 ppm for the same species (see the ESI‡). Broader signals in the same area were attributed to the propagating 1,2-oxaphosphetan-2-ium end units of growing oligomers 11n. A broad signal group from 50 to 58 ppm is assigned to the various open chain phosphane oxide units of the oligomers 11n.
While knowing that 1,2-oxaphosphetanes are moderately strained (ring strain energy of the parent compound, RSE = 19.09 kcal mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27), DFT calculations on the Arbuzov reaction pathway of 6 were performed and, hence, the reaction of the model (S)-2-methyl-1,2-oxaphosphetane 6* with methyl iodide (8d) was computed. The primary step is the exergonic P-methylation, affording 2,2-dimethyl-1,2-oxaphosphetan-2-ium iodide 9d* (Scheme 5 and Fig. 5), which led to a significant increase of the RSE from 18.66 to 26.24 kcal mol−1 at the (gas phase) working level of theory,37 in agreement with the enlarged Lagrangian of the kinetic energy density per electron computed at the ring critical point38 (G/ρ = 1.1168 and 1.1392 a.u. for 6* and 9*, respectively), thus paralleling the observed tendency of oxaphosphiranes.39 The Arbuzov-type reaction of the iodide counter anion in 9d* is largely facilitated due to the RSE relief, thus proceeding exergonically over a low energy barrier (Fig. 5) and affording β-iodoethyl phosphane oxide 10d*. In the case of using methyl triflate (8e) as the methylating agent, the energy barrier is slightly lower (ΔGrel = 23.46 kcal mol−1) and the resulting 1,2-oxaphosphetan-2-ium triflate 9e* (ΔGrel = –37.35 kcal mol−1) is much more stable than the iodide analogue 9d*. This is due to the presence of a rather strong anion⋯P bonding interaction with the triflate O− centre in 9e* (dP⋯O = 2.385 Å) than with I− in 9d* (dP⋯I = 3.314 Å). The former highlights the ambiguity of the WCA character of the triflate anion and its inability to promote Arbuzov-type ring opening in 9e*.
27), DFT calculations on the Arbuzov reaction pathway of 6 were performed and, hence, the reaction of the model (S)-2-methyl-1,2-oxaphosphetane 6* with methyl iodide (8d) was computed. The primary step is the exergonic P-methylation, affording 2,2-dimethyl-1,2-oxaphosphetan-2-ium iodide 9d* (Scheme 5 and Fig. 5), which led to a significant increase of the RSE from 18.66 to 26.24 kcal mol−1 at the (gas phase) working level of theory,37 in agreement with the enlarged Lagrangian of the kinetic energy density per electron computed at the ring critical point38 (G/ρ = 1.1168 and 1.1392 a.u. for 6* and 9*, respectively), thus paralleling the observed tendency of oxaphosphiranes.39 The Arbuzov-type reaction of the iodide counter anion in 9d* is largely facilitated due to the RSE relief, thus proceeding exergonically over a low energy barrier (Fig. 5) and affording β-iodoethyl phosphane oxide 10d*. In the case of using methyl triflate (8e) as the methylating agent, the energy barrier is slightly lower (ΔGrel = 23.46 kcal mol−1) and the resulting 1,2-oxaphosphetan-2-ium triflate 9e* (ΔGrel = –37.35 kcal mol−1) is much more stable than the iodide analogue 9d*. This is due to the presence of a rather strong anion⋯P bonding interaction with the triflate O− centre in 9e* (dP⋯O = 2.385 Å) than with I− in 9d* (dP⋯I = 3.314 Å). The former highlights the ambiguity of the WCA character of the triflate anion and its inability to promote Arbuzov-type ring opening in 9e*.
 | ||
| Scheme 5 Reaction of 6* with methyl bromoacetate 8a. | ||
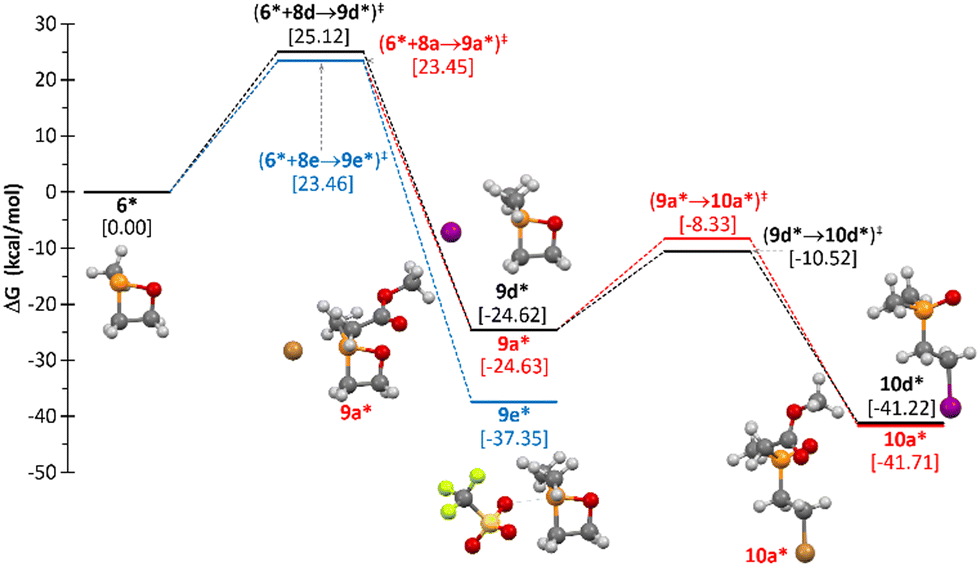 | ||
| Fig. 5 Computed (CPCMsolv/PWPB95-D3/def2-QZVPP//CPCMsolv/PBEh-3c) Gibbs energy profile for the reaction of model 6* with 8a, d, and e. Solvent: THF for reaction with 8a and d but CH2Cl2 for 8e. | ||
Similarly, the reaction of 6 with methyl bromoacetate 8a occurred selectively giving a β-bromoethyl phosphane oxide, like compound 10a* in the computed model reaction (Fig. 5 and Scheme 5). Intermediate 9a* also displays a remarkable pnictogen bonding interaction with the Br− formal anion (dP⋯Br = 2.665 Å), but the value is well below the sum of the van-der-Waals radii (1.80 + 1.85 = 3.65 Å). However, α-halocarbonyl compounds feature a second electrophilic site at the carbonyl C atom which can allow for the formation of the Perkow product, in general,40,41 that was shown to proceed through initial formation of a σ5λ5-oxaphosphirane intermediate.42 In the case of the reaction of 6* with methyl bromoacetate (8d), the alternative Perkow route was investigated computationally (see the ESI‡) which turned out to be both kinetically and thermodynamically disfavoured compared to the Arbuzov route. The kinetic preference for the Michaelis–Arbuzov pathway can be explained in terms of most favoured frontier molecular orbital (FMO) interactions (i.e. the phosphorus lone pair of 6 and the σ*(C–Br)-orbital of 8a, see the ESI‡). For 8c, the only observed product is again the thermodynamically most stable Arbuzov product 10c.
In total, reactions of a C,C-unsubstituted 1,2σ3λ3-oxaphosphetane with various organic bromo and iodo derivatives were studied, including methyl triflate, to probe the effect of the counter anion. While bromo and iodo derivatives led cleanly to Arbuzov-type products, the β-halo phosphane oxides, methyl triflate led to a mixture of low-weight oligomers. In the latter case, VT-NMR studies provided evidence for the first 1,2-oxaphosphetan-2-ium salt formed at low temperature, which was also supported by DFT calculations.
FG carried out experiments and evaluated analytical data. GS is responsible for the crystallographic data. RS provided concept and the supervised the investigation. FG, AEF and RS wrote the manuscript. The ESI‡ was prepared by FG, AEF and RS.
We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft and the University of Murcia for the computational resources of Servicio de Cálculo Científico (SCC).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815 CrossRef CAS PubMed.
- J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150 CrossRef CAS PubMed.
- R. Klein and F. R. Wurm, Macromol. Rapid Commun., 2015, 36, 1147 CrossRef CAS PubMed.
- A. Marinetti and D. Carmichael, Chem. Rev., 2002, 102, 201 CrossRef CAS PubMed.
- W. Zhao, P. K. Yan and A. T. Radosevich, J. Am. Chem. Soc., 2015, 137, 616 CrossRef CAS PubMed.
- K. D. Reichl, N. L. Dunn, N. J. Fastuca and A. T. Radosevich, J. Am. Chem. Soc., 2015, 137, 5292 CrossRef CAS PubMed.
- T. V. Nykaza, T. S. Harrison, A. Ghosh, R. A. Putnik and A. T. Radosevich, J. Am. Chem. Soc., 2017, 139, 6839 CrossRef CAS PubMed.
- L. Longwitz, A. Spannenberg and T. Werner, ACS Catal., 2019, 9, 9237 CrossRef CAS.
- L. Longwitz and T. Werner, Pure Appl. Chem., 2019, 91, 95 CrossRef CAS.
- L. Longwitz and T. Werner, Angew. Chem., Int. Ed., 2020, 59, 2760 CrossRef CAS PubMed.
- B. Kaboudin, H. Haghighat and T. Yokomatsu, Synthesis, 2011, 3185 CrossRef CAS.
- G. Wittig and G. Geissler, Justus Liebigs Ann. Chem., 1953, 580, 44 CrossRef CAS.
- A. Hinz, R. Labbow, C. Rennick, A. Schulz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2017, 56, 3911 CrossRef CAS PubMed.
- G. Wittig and U. Schöllkopf, Chem. Ber., 1954, 87, 1318 CrossRef.
- E. Vedejs, G. P. Meier and K. A. J. Snoble, J. Am. Chem. Soc., 1981, 103, 2823 CrossRef CAS.
- G. Wittig and W. Haag, Chem. Ber., 1955, 88, 1654 CrossRef CAS.
- A. Espinosa Ferao, ChemPlusChem, 2023, e202300474 CrossRef PubMed.
- A. Espinosa Ferao, Inorg. Chem., 2018, 57, 8058 CrossRef CAS PubMed.
- U. Dieckbreder, E. Lork, G.-V. Röschenthaler and A. A. Kolomeitsev, Heteroat. Chem., 1996, 7, 281 CrossRef CAS.
- M. Hamaguchi, Y. Iyama, E. Mochizuki and T. Oshima, Tetrahedron Lett., 2005, 46, 8949 CrossRef CAS.
- T. Kawashima, K. Kato and R. Okazaki, Angew. Chem., Int. Ed. Engl., 1993, 32, 869 CrossRef.
- Mazhar-ul-Haque, C. N. Caughlan, F. Ramirez, J. F. Pilot and C. P. Smith, J. Am. Chem. Soc., 1971, 93, 5229 CrossRef CAS.
- E. N. Dianova, E. Y. Zabotina, I. Z. Akhmethkhaova and Y. D. Samuilov, Zh. Obshch. Khim., 1991, 1063 CAS.
- A. W. Kyri, G. Schnakenburg and R. Streubel, Chem. Commun., 2016, 52, 8593 RSC.
- A. W. Kyri, Investigations on 1,2-Oxaphosphetane Complexes. PhD Thesis, University of Bonn, Germany, 2017.
- A. W. Kyri, V. Nesterov, G. Schnakenburg and R. Streubel, Angew. Chem., Int. Ed., 2014, 53, 10809 CrossRef CAS PubMed.
- A. W. Kyri, F. Gleim, A. García Alcaraz, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Chem. Commun., 2018, 54, 7123 RSC.
- A. Espinosa Ferao, B. Deschamps and F. Mathey, Bull. Soc. Chim. Fr., 1993, 695 Search PubMed.
- F. Gleim, A. García Alcaraz, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Molecules, 2022, 27, 3345 CrossRef CAS PubMed.
- B. A. Arbusow, Pure Appl. Chem., 1964, 9, 307 CrossRef.
- A. Michaelis and R. Kähne, Ber. Dtsch. Chem. Ges., 1898, 31, 1048 CrossRef.
- A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415 CrossRef CAS.
- S. Kobayashi, M. Suzuki and T. Saegusa, Macromolecules, 1984, 17, 107 CrossRef CAS.
- F. Mathey and F. Mercier, J. Chem. Soc., Chem. Commun., 1980, 0, 191 RSC.
- G. A. Lawrance, Chem. Rev., 1986, 86, 17 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed.
- RSE 20.19 (6*) and 27.62 kcal mol−1 (9*), at the standard DLPNO-CCSD(T)/def2-QZVPP//B3LYP-D4/def2-TZVP level of theory.
- A. Espinosa Ferao, Tetrahedron Lett., 2016, 57, 5616 CrossRef CAS.
- A. Espinosa Ferao, A. Rey Planells and R. Streubel, Eur. J. Inorg. Chem., 2021, 348 CrossRef CAS.
- W. Perkow, K. Ullerich and F. Meyer, Sci. Nat., 1952, 39, 353 CrossRef.
- W. Perkow, Chem. Ber., 1954, 87, 755 CrossRef CAS.
- A. Espinosa Ferao, J. Phys. Chem. A, 2017, 121, 6517 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Dr Hansjörg Grützmacher on the occasion of his 65th birthday and in memory of Prof. Dr Edgar Niecke, one of the pioneers of modern chemistry of small P-heterocycles. |
| ‡ Electronic supplementary information (ESI) available: Experimental protocols, NMR and theoretical data. CCDC 2290099–2290104. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc00254g |
| This journal is © The Royal Society of Chemistry 2024 |